«Забытая» иммунология эпидемических, инфекционных и поствакцинальных процессов

О серьезных пробелах в знаниях об иммунологии эпидемических, инфекционных и поствакцинальных процессов говорит то, что до настоящего времени распространение многих опасных инфекционных болезней не удается контролировать с помощью вакцинации, а результативность самих вакцинаций вызывает все больше вопросов у врачей. В работе рассмотрены феномены антигенного импринтинга и антителозависимого усиления инфекции. Оба феномена обнаружены более 50 лет назад, однако в российских руководствах для врачей их описание отсутствует. Показано, что иммунология, так как она преподается в медицинских вузах, носит чрезмерно упрощенный характер. Тот объем знаний, которые получают в этой области врачи, выгоден лишь недобросовестным производителям устаревших вакцин. Оба «забытых» иммунологических феномена играют важную роль в эпидемических процессах и в определенных ситуациях способны усиливать инфекционный процесс у реконвалесцентов и ранее вакцинированных пациентов. Они также являются основным препятствием в создании вакцин против возбудителей гриппа, ретровирусных инфекций, лихорадок Денге, Марбург, Эбола, гепатита С, энцефалитов Западного Нила и долины Мюррей, малярии и некоторых других. Их игнорирование при подготовке медицинских специалистов в такой сфере, как борьба с инфекционными болезнями, создает благоприятные условия для злоупотреблений фармацевтических компаний, тормозит развитие отечественной эпидемиологии и иммунологии, и отдает приоритет новых открытий зарубежным исследователям.
Библиографическое описание: Супотницкий М.В. «Забытая» иммунология эпидемических, инфекционных и поствакцинальных процессов // Новости медицины и фармации. — 2014. — № 9–10. — С. 19–23; № 11–12. — С. 16–20.
Распространенные учебные руководства для медицинских работников [1–7] содержат типичные представления об иммунитете, из которых не следует, почему до настоящего времени не создано вакцин, позволяющих контролировать распространение многих опасных инфекций. Цель настоящей работы — обратить внимание врачей на пробелы в знаниях об иммунологии эпидемических, инфекционных и поствакцинальных процессов, получаемых ими в медицинских вузах. Рассмотрена роль в этих процессах феноменов антителозависимого усиления инфекции и антигенного импринтинга[1].
1.Формирование типичных представлений об иммунитете
Сложившиеся в конце XIX в. представления о специфическом иммунитете отражают взгляды Э. Дженнера и Л. Пастера, априори предполагавших, что первый контакт с возбудителем инфекционной болезни создает у человека невосприимчивость (иммунитет) к повторному заражению [11].
Теория опсонинов, разработанная А.Е. Райтом и С. Дугласом в 1903 г. [12], примирила враждующие между собой фагоцитарную (И.И. Мечников) и гуморальную (Р. Кох, П. Эрлих) теории иммунитета и дала толчок к ее дальнейшему развитию на основе представлений о кооперации клеточно-гуморальных иммунологических реакций. В последующие годы были описаны и апробированы иммунологические реакции и тесты с фагоцитирующими клетками и специфическими антителами, уточнялся механизм их взаимодействия с антигенами. В 1948 г. А. Фагреус доказала, что антитела синтезируются плазмоцитами. Иммунологическая роль Т- и В-лимфоцитов установлена в 1960-х гг., когда было показано превращение В-клеток в плазмоциты под влиянием антигенов и образование из недифференцированных Т-клеток несколько субпопуляций, синтезирующих специфические к антигену антитела. В 1966 г. открыты цитокины Т-лимфоцитов, обусловливающие кооперацию (взаимодействие) иммунокомпетентных клеток. Сформулированная в 1964 г. Н. Йерне и Ф. Бернетом клонально-селекционная теория иммунитета дала ученым понимание того, каким образом специфические антитела могут накапливаться в достаточно высокой концентрации, чтобы эффективно блокировать инфекционный процесс. Подобный же механизм установлен и при формировании клоноспецифических Т-клеток [17].
В настоящее время типичная схема иммунного ответа на возбудитель инфекционной болезни выглядит следующим образом. Макрофаг поглощает (фагоцитирует) патогенный микроорганизм (бактерия, вирус), инактивирует его и презентирует Т- и В-лимфоцитам. Ввиду различий рецепторного аппарата В-клетки реагируют с одними детерминантами, Т-клетки — с другими. Получив информацию об антигене от антиген-презентирующих клеток (макрофаги, несущие на внешней мембране антигены), Т-хелперы с помощью иммуноцитокинов передают сигнал, усиливающий пролиферацию Т- и В-лимфоцитов определенных клонов[2]. В-лимфоциты дифференцируются до плазмоцитов, а Т-хелперы превращаются в Т-киллеры (Т-эффекторы). Плазмоциты синтезируют специфические антитела, участвующие в иммунном ответе в трех формах — нейтрализации, опсонизации и активации системы комплемента (гуморальный иммунный ответ); Т-киллеры разрушают клетки-мишени при непосредственном контакте (цитотоксический или клеточный иммунный ответ). После первичного контакта с антигеном остаются клоны Т- и В-клеток памяти, сохраняющие информацию о нем много лет. При вторичном попадании этого антигена в организм человека они рекрутируют специфические лимфоциты, происходит стимуляция этих клонов, и клетки памяти начинают интенсивно размножаться[3]. В-клетки переходят в плазмоциты, продуцирующие антитела нужной специфичности. Т-клетки обеспечивают клеточную форму защиты (субпопуляции цитотоксических Т-клеток и Т-клеток воспаления) и участвуют в формировании гуморального иммунитета — хелперные Т-клетки [4, 5, 17].
В соответствии с такой схемой гуморальные и клеточные реакции, развивающиеся в ответ на введение вакцины или развитие инфекционного процесса, могут иметь исключительно защитный характер. Поэтому любой препарат, полученный на основе антигенов какого-то возбудителя инфекционной болезни и вызывающий образование специфических к этим антигенам антител, можно назвать вакциной, а любую массовую вакцинацию легко представить всечеловеческим благом.
2.Феномен антигенного импринтинга[4]
В 1953 г. Davenport et al. [18] неожиданно для себя обнаружили, что в сыворотке крови людей старше 28 лет, переболевших гриппом до проводимых в начале 1950–х гг. массовых вакцинаций, низкие титры антител к вирусу серотипа А (H1N1)[5], использованному при приготовлении вакцины, но повышенное содержание антител к вирусу гриппа, циркулировавшему ранее. Наибольшее количество таких людей приходится на возрастную группу 35–38 лет, пережившую пандемию гриппа «испанка» в 1918 г. Аналогичные результаты были получены в отношении вируса гриппа серотипа B и его антигенных вариантов [19]. Для объяснения иммунологического феномена Davenport et al. [18] предположили, что во время первого инфицирования вирусом гриппа еще в детском возрасте иммунная система ориентируется на некий доминантный антиген среди циркулирующих штаммов вируса. Последующее экспонирование к вирусам гриппа, антигенно связанным с предыдущим, вызывает подъем уровня антител не на их антигены, а на штамм, вызвавший первую инфекцию. Это наблюдение было кратко резюмировано Francis [19] в виде «доктрины первичного антигенного греха» (the doctrine of original antigenic sin).
Установление природы феномена антигенного импринтинга. Davenport и Hennessy [20] для определения границ феномена провели вакцинацию моновалентными вакцинами, содержащими инактивированные штаммы различных антигенных вариантов (сероподтипов) вируса гриппа А, циркулировавших среди людей за последние 30 лет. Среди них вирус свиного гриппа (Hsw1N1; swine influenza) — циркулировал во время пандемии «испанки» 1918 г. и некоторое время позже, вирус гриппа А (H0N1)[6] — вызывал вспышки гриппа с начала 1930–х гг. до 1943 г., вирус гриппа А-prime (H1N1, influenza A-prime) — доминировал в циркуляции среди людей с 1946 г. до начала 1950–х гг.
Вакцинация была проведена в группе детей, которые были вовлечены в вспышки гриппа, вызванные вирусом гриппа сероподтипа A-prime, в группах взрослых (рекруты), детьми переживших вспышки гриппа А, и взрослых людей старше 30 лет. У детей высокие титры антител отмечены на вакцину на основе вируса гриппа A-prime (H1N1), у рекрутов — на вакцину против вируса гриппа А (H0N1), у людей старше 30 лет — на вакцину на основе вируса свиного гриппа (Hsw1N1). У некоторых волонтеров двух последних групп были обнаружены антитела к вирусам гриппа A-prime (H1N1), свидетельствующие о ранее перенесенной инфекции. Реакция человека на введение моновалентных вакцин оказалась типоспецифической. Антитела к вирусу гриппа A-prime, полученные в результате вакцинации детей по гриппу A или свиному гриппу, не вступали в перекрестные реакции с вирусами гриппа A или свиного гриппа. Такие же результаты получены в группах рекрутов (антитела к вирусу гриппа A) и людей старше 30 лет (антитела к вирусу свиного гриппа). Этим изящным экспериментом Davenport и Hennessy [20] подтвердили ранее полученные ими данные [18], говорящие в пользу того, что иммунная система человека при сходстве антигенов может реагировать на тот, с которым она «столкнулась» впервые.
К концу 1950-х гг. предположение Davenport и Hennessy [20] подтверждено эпидемиологическими исследованиями. Было установлено, что антитела к различным типам вируса гриппа накапливаются в течение всей жизни человека, однако после эпидемических вспышек болезни титр антител бывает наивысшим к тому типу вируса, который обусловил первое заболевание гриппом в раннем детстве [14]. Francis (1959) обнаружил следующую закономерность распределения антител к сероподтипам вируса гриппа типа А среди возрастных групп населения США (табл. 1).
Таблица 1. Распределение антител к сероподтипам вируса гриппа типа А в сыворотке людей из разных возрастных групп населения США*
|
Разновидность вируса (год появления в США) |
Возраст пациента |
|
А2 (1957) |
Начиная с 70–80 лет и старше |
|
Свиной (1931) |
–‘'– 35–40 –‘'– |
|
А (1934) |
–‘'– 15–35 –‘'– |
|
А1 (1947) |
–‘'– 1–10 –‘'– |
*Цит. по [14].
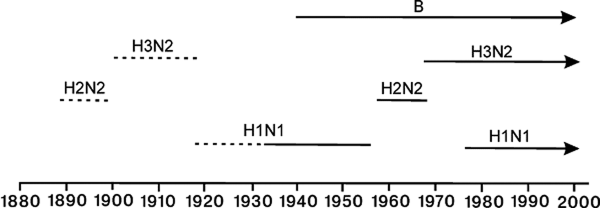
В конце 1950-х гг. эпидемическая ситуация по гриппу изменилась. Вирусы сероподтипов Hsw1N1, H0N1, H1N1 сменил вирус сероподтипа H2N2 (пандемия азиатского гриппа 1957 и 1959 гг.), затем в циркуляции среди людей появился вирус сероподтипа H3N2 (пандемия гонконгского гриппа 1968–1970 гг.) (рис. 1).
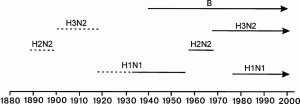 Рисунок 1. Пандемические циклы вируса гриппа типа А человека. По [21]
Рисунок 1. Пандемические циклы вируса гриппа типа А человека. По [21]
Феномен антигенного импринтинга в 1960-е гг. не только не вызывал сомнения у эпидемиологов и иммунологов, но и использовался ими для разработки методологии археологической серологии. Методология основывалась на определении возрастного распределения антител к различным антигенным вариантам вирусов А и В. Различия в распределении антител среди возрастных групп населения связывали с возникновением анамнестических реакций на вирусы с близкими по структуре антигенами, ранее вызвавшими у человека заболевание гриппом. Благодаря такому подходу установлено, что вирусы гриппа, сходные с А2N2 и В, циркулировавшие среди людей в начале 1960–х гг., вызывали эпидемии гриппа в 1880–1890-х гг. Для распознания по серологическим показателям истинного сероварианта возбудителя гриппа обычно обследовали однородный контингент по возрасту (пионерские лагеря, общежития, воинские части) [14].
Появившиеся в циркуляции среди людей вирусы гриппа сероподтипов H2N2 и H3N2 давали собственные анамнестические ответы иммунной системы. Marine и Thomas [22], проводя серологические исследования среди людей различных возрастных групп, вакцинированных моновалентными инактивированными вакцинами на основе вирусов гриппа серотипа А различных антигенных вариантов (H1N1 и H0N1, H2N2, H3N2), установили, что антигенный импринтинг наблюдается в пределах одного антигенного варианта вируса. У людей, перенесших первую гриппозную инфекцию, вызванную вирусами сероподтипов H1N1и H0N1, была анамнестическая реакция (высокие титры антител) на вакцинацию вакцинами, полученными на основе штаммов вирусов этих сероподтипов, но не H2N2 и H3N2, и наоборот.
Эффект антигенного импринтинга проявлялся тем интенсивнее, чем больше времени проходило от момента первого контакта иммунной системы с возбудителем гриппа. Masurel и Drescher [23] в опытах на хорьках, последовательно инфицированных с интервалами в три недели разными штаммами вируса гриппа серотипа А (H1N1, Hsw1N1, H0N1, H2N2, H3N2), установили, что вторичное инфицирование может приводить к появлению HCR-антител, т.е. антител с высокой перекрестной активностью (antibodies highly cross-reacting — HCR antibodies) по отношению к штаммам, антигенно тесно связанным по гемагглютинину (НА) с теми, что вызвали первый инфекционный процесс. При заражении вирусом гриппа через трехнедельные интервалы антител, специфичных к штамму вируса, вызвавшему первый случай инфекции, не обнаруживали. Однако, когда интервал между заражениями увеличивали до 4–5 мес., наблюдался феномен антигенного импринтинга, а HCR-антитела не обнаруживали. Следовательно, образование HCR-антител и антигенный импринтинг — это разные иммунологические феномены.
В 1979 г. при анализе статистики заболеваемости населения гриппом обнаружено, что люди, родившиеся до 1956 г., легко перенесли пандемию русского гриппа (1977–1978 гг.). Подавляющее число заболевших приходилось на людей в возрасте до 20 лет, т.е. на ту часть населения, которая не имела контакта с вирусами гриппа серотипа H1N1, вышедшими из циркуляции более 20 лет тому назад. Напротив, лица старше 30 лет составили только 20 % больных, хотя их доля в общей численности населения превышает 50 %, т.е. с учетом низкой заболеваемости в эту эпидемию вообще люди зрелого и пожилого возраста, имевшие в прошлом контакт с вирусами гриппа H1N1, болели значительно меньше, чем люди более молодых возрастных групп [24]. Данный феномен наблюдался во всех странах, где велся учет заболевших гриппом, и был объяснен антигенным импринтингом. В экспериментах, выполненных на крысах, установлено отсутствие влияния на анамнестические ответы иммунной системы на вирус сероподтипа H1N1, последующего инфицирования вирусом гриппа других сероподтипов (H2N2, H3N2) [25].
Marine и Thomas [22] подтвердили феномен антигенного импринтинга в масштабном исследовании, выполненном на 687 добровольцах разных возрастов, перенесших грипп во время различных пандемий. Добровольцев вакцинировали живыми моновакцинами разных серотипов и изучали анамнестические ответы иммунной системы. В этом же году Couch et al. [26] обнаружили, что после вакцинации инактивированной гриппозной вакциной, полученной на основе штамма вируса A/Scotland/74, в сыворотке 82 % вакцинированных людей обнаруживались антитела к вирусу A/HongKong/68, с которым они сталкивались во время предыдущих вспышек гриппа. Только в сыворотке 46 % из них были обнаружены низкие уровни антител к вакцинному штамму A/Scotland/74.
Но в практике вакцинаций по гриппу феномен антигенного импринтинга подтверждался не всегда (см., например, [27]). Границы изменчивости вируса гриппа в пределах его сероподтипов, при которых этот феномен возможен, пытались в 1999 г. смоделировать Smith et al. [28]. По их данным, чем больше антигенное сходство между штаммами вируса гриппа, использованными для приготовления вакцины, которая применялась для вакцинации, и штаммами вируса, вызвавшего вспышку гриппа, или антигеном вируса, использованного для повторной вакцинации, тем больше вероятность развития феномена антигенного импринтинга и тяжелого течения болезни у инфицированного пациента. При полной антигенной идентичности вирусов антигенный импринтинг невозможен. Но конкретных величин антенного различия вирусов, при котором он может возникнуть или быть исключенным, они не привели.
Тогда же было обнаружено, что явление антигенного импринтинга может наблюдаться не только при гуморальном, но и при клеточном иммунном ответе на возбудители инфекционных болезней. При повторном реагировании на мутировавшие антигены вируса лимфоцитарного хорио–менингита (lymphocytic choriomeningitis virus — LCMV), узнаваемые цитотоксическими Т-клетками, цитотоксический ответ происходил преимущественно в отношении того антигенного варианта вируса, с которым иммунная система человека взаимодействовала первично [29]. В 2010 г. аналогичная роль Т-клеточных ответов иммунной системы человека описана при лихорадке Денге [30]. Антигенный импринтинг наиболее опасен при развитии повторной инфекции тогда, когда в результате В- и Т-клеточных ответов образуются низкоавидные перекрестно реагирующие антитела на доминирующие антигенные эпитопы, как, например, это происходит в отношении эпитопов оболочечного белка Е вируса Денге [31]. Такие антитела, образующиеся на ранней стадии повторной инфекции, являются причиной развития другого малоизученного иммунологического феномена — антителозависимого усиления инфекции [32]
В 1990-х гг. изменилась ситуация в самой эпидемиологии. Началось время глобальных информационных проектов фармацевтических корпораций, имеющих целью сверхприбыль от коммерческой реализации вакцин. Представления об эпидемиологии и иммунологии инфекционных болезней упростились. Теперь эпидемии уже не были результатом сложных природных и социальных процессов, а возникали из-за появления нового вируса, что автоматически предполагало новую вакцину, массовую вакцинацию и бюджетное финансирование ее проведения. Ответы со стороны иммунной системы на вакцину либо на возбудитель инфекционной болезни в интересах производителей вакцин стали рассматриваться исключительно как защитные. Феномен антигенного импринтинга исчез из руководств и учебников, и современным российским врачам, в отличие от их советских коллег 1960-х гг., он неизвестен.
Антигенный импринтинг в период пандемии свиного гриппа в 2009 г. «Забытый» иммунологический феномен вспомнили исследователи, не связанные с вакцинным бизнесом, когда стали изучать последствия массовых вакцинаций, навязанных населению фармацевтическими корпорациями под предлогом предотвращения перехода пандемии свиного гриппа в «испанку»[7]. В 2009 г. Kim et al. [33] подтвердили возможность развития феномена антигенного импринтинга в экспериментах на мышах, используя штаммы A/PR/8/34 (PR8) и A/FM/1/47 (FM1) вируса сероподтипа H1N1. Аминокислотная последовательность HA обоих штаммов была идентична на 92 %.
Также они показали, что если проводить последовательную вакцинацию мышей инактивированными вакцинами, полученными на основе разных штаммов вируса гриппа (PR8 и FM1), то при последующем заражении адаптированным штаммом FM1 мыши оказываются менее защищенными от вируса, чем после иммунизации только одним инактивированным FM1. Титр вируса гриппа в легких мышей, вакцинированных сначала PR8, а затем FM1, был в 46 раз выше, чем у мышей, вакцинированных только инактивированным FM1. Мыши, вакцинированные сначала инактивированной вакциной, затем живой, демонстрировали выраженный антигенный импринтинг. Последующее инфицирование животных вирулентным штаммом вируса вызывало у них слабый ответ нейтрализующих антител на этот вирус. Индукция феномена антигенного импринтинга не зависела от введенной дозы вирусов (0,01 или 0,1 LD50) или последовательности, в которой они были введены экспериментальному животному.
Choi et al. [34] обнаружили, что 18–20-летние студенты, ранее многократно вакцинированные вакцинами, предназначенными для сезонной вакцинации по гриппу, реагировали на гриппозную вакцину, разработанную для противодействия распространению пандемического вируса сероподтипа рH1N1 (pandemic H1N1 2009; pH1N1), значительно слабее, чем те студенты, кого раньше не вакцинировали. Однако выяснить, какая вакцинация стала причиной антигенного импринтинга, исследователям не удалось, так как за последние 15 лет в состав вакцин для сезонной вакцинации включалось шесть различных штаммов (!) вируса гриппа сероподтипа H1N1. Установлено только то, что это не была комбинированная вакцина, включающая вирус A/Brisbane/59/2007(H1N1), которая была использована три месяца назад для вакцинации населения. Она не создавала значительного перекрестного защитного эффекта по отношению к вирусу pH1N1.
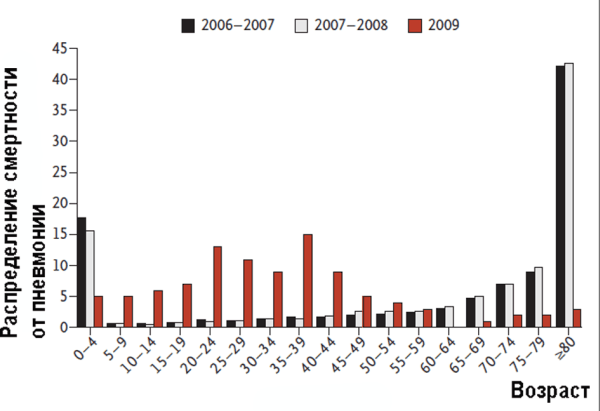
Анализ заболеваемости в разных возрастных группах населения во время глобальной активизации вируса рH1N1 в 2009 г. дал тот же результат, что и подобные анализы заболеваемости, проведенные в начале 1950-х гг. и после пандемии русского гриппа в конце 1970-х гг. У людей, родившихся до 1957 г., антигенный импринтинг стал причиной высоких титров вирус-нейтрализующих антител, вырабатывающихся как в ответ на вакцинацию, так и на гриппозную инфекцию. В других же возрастных группах антигенный импринтинг повышал смертность заболевших [35–38] (рис. 2).
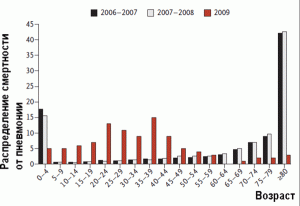 Рисунок 2. Распределение смертности от пневмонии по разным возрастным группам в 2009 г. в Мехико на фоне пандемии гриппа. Приводится сравнение с аналогичными показателями во время сезонных вспышек гриппа в период 2006–2008 гг. По [37]
Рисунок 2. Распределение смертности от пневмонии по разным возрастным группам в 2009 г. в Мехико на фоне пандемии гриппа. Приводится сравнение с аналогичными показателями во время сезонных вспышек гриппа в период 2006–2008 гг. По [37]
Четыре эпидемиологических исследования распространения вируса пандемического гриппа рH1N1, выполненные в Британской Колумбии в 2009 г., показали повышенный риск его развития у лиц, ранее вакцинированных тривалентной инактивированной гриппозной вакциной (trivalent inactivated influenza vaccine, TIV), применяемой для сезонной профилактики гриппа. Авторы связывают повышенный риск развития гриппа у вакцинированных людей с феноменами антигенного импринтинга, антителозависимого усиления инфекции и с другими, еще не известными факторами, на необходимость изучения которых они обращают внимание исследователей [39, 40].
После предшествующих многократных вакцинаций и перенесенных в прошлом заболеваний гриппом антигенный импринтинг приводит к тому, что в сыворотке крови циркулируют специфические низкоавидные антитела, перекрестнореагирующие с вирусами гриппа, но не обладающие протективным действием. Вспышки свиного гриппа 2009 г. внесли ясность и в их роль в патогенезе гриппозной инфекции.
По данным Monsalvo et al. [41], у умерших пациентов среднего возраста и тех, у кого грипп имел тяжелое течение, специфические низкоавидные антитела (IgG) формировали иммунные комплексы с вирусом, оседавшие в легочной ткани и вызывавшие отек легких, перибронхиолярную мононуклеарную клеточную инфильтрацию и как результат — гипоксемию. Чем выше был титр таких антигриппозных антител, тем тяжелее протекала болезнь. У пациентов не обнаруживали антител, нейтрализующих pH1N1, и находили вирус гриппа в легочной ткани в высоких титрах. По результатам их исследования в Аргентине прекратили вакцинацию детей против гриппа до 2010 г.
Reichert et al. [38] обнаружили один из механизмов изменений в антигенной структуре НА вируса гриппа, приводящих к развитию антигенного импринтинга при повторном взаимодействии вируса с иммунной системой человека. По их данным, HA вируса рH1N1 тесно связан с HA вируса, вызвавшего пандемию гриппа «испанки» в 1918 г., и вирусов, циркулировавших в период с 1930-х по 1943 г. Эволюция вирусов сероподтипа H1N1, циркулировавших в популяциях людей в 1940–50-е гг. и после его возвращения в 1977 г., происходила через гликозилирование HA (т.е. присоединение остатков сахаров к HA) (рис. 3).
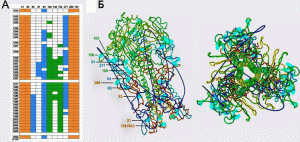 Рисунок 3. Гликозилирование гемагглютинина вируса гриппа сероподтипа H1N1 за период 1918–2009 гг. A. Гликозилирование HA вируса H1N1 за период 1918–2009 гг. — бежевым цветом показаны потенциальные сайты гликозилирования в домене слияния в позициях 21, 33, 289 и 154 (HA2). Синий цвет соответствует участкам гликозилирования в позициях 63, 81, 94 и 271 в домене рудиментарной эстеразы (vestigial esterase). Зеленым цветом показаны участки гликозилирования в положениях 129 (или 131), 156 и 163 на глобулярной головке HA вблизи рецептор-связывающего сайта вируса. Б. Структурное соответствие HA-тримера кристалической структуры вируса H1N1, вызвавшего пандемию 1918 г. HA-тримерам вирусов сероподтипа H1N1, появившихся в циркуляции среди людей позже 1918 г. Сайты гликозилирования наложены поверх изображения в цветах, показанных на панели А. По [38]
Рисунок 3. Гликозилирование гемагглютинина вируса гриппа сероподтипа H1N1 за период 1918–2009 гг. A. Гликозилирование HA вируса H1N1 за период 1918–2009 гг. — бежевым цветом показаны потенциальные сайты гликозилирования в домене слияния в позициях 21, 33, 289 и 154 (HA2). Синий цвет соответствует участкам гликозилирования в позициях 63, 81, 94 и 271 в домене рудиментарной эстеразы (vestigial esterase). Зеленым цветом показаны участки гликозилирования в положениях 129 (или 131), 156 и 163 на глобулярной головке HA вблизи рецептор-связывающего сайта вируса. Б. Структурное соответствие HA-тримера кристалической структуры вируса H1N1, вызвавшего пандемию 1918 г. HA-тримерам вирусов сероподтипа H1N1, появившихся в циркуляции среди людей позже 1918 г. Сайты гликозилирования наложены поверх изображения в цветах, показанных на панели А. По [38]
Гликозилирование HA сформировало то антигенное разнообразие среди вирусов гриппа, вызывающих сезонные вспышки болезни, которое проявилось антигенным импринтингом в отдельных возрастных группах после появления в циркуляции вируса pH1N1.
Специфичность антигенного импринтинга, проявившаяся защитным эффектом в старших возрастных группах населения, и сравнительные данные по гликозилированию HA вирусов гриппа (рис. 3) свидетельствуют в пользу того, что вирус pH1N1 идентичен вирусу, преобладавшему в циркуляции среди людей в первой трети ХХ в. Тогда возникают следующие вопросы:
1)где и как вирус pH1N1поддерживался в природе почти 80 лет без гликозилирования HA?
2)каковы механизмы его глобального вовлечения в эпидемические процессы?
3)почему в этот раз он не вызвал смертельно опасную пандемию гриппа — «испанку»?
Антигенный импринтинг при ВИЧ-инфекции. Феномен показан:
1)при исследовании защитного действия анти-ВИЧ-вакцин;
2)инфекционном процессе, вызванном ВИЧ [42].
Первыми на антигенный импринтинг при разработке ВИЧ-вакцин натолкнулись Nara et al. [43]. Они пытались расширить иммунный ответ на антигены ВИЧ в отношении вирусов различного географического происхождения. Введя шимпанзе гликопротеид gp120, полученный из штамма ВИЧ-1 IIIB, и проведя через 175 сут. повторную вакцинацию gp120, выделенным из штамма ВИЧ-1 RF, имеющего другое географическое происхождение, они неожиданно для себя обнаружили рост титров антител к gp120 штамма IIIB и отсутствие защитного эффекта при заражении животных ВИЧ-1 RF. Проведенный ими ретроспективный анализ научной литературы показал, что феномен OAS уже был описан для других ретровирусных инфекций, в частности вызываемых вирусом висны у овец [44] и вирусом инфекционной анемии у лошадей [45] (рис. 4).
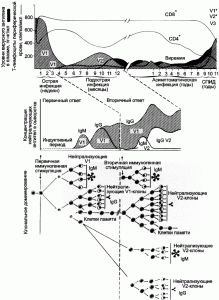 Рисунок 4. Модель феномена «первичного антигенного греха» при ВИЧ-инфекции у людей, вызванного презентацией иммунной системе gp120. Заштрихованная область, обозначенная как V1, соответствует вирусной нагрузке, образовавшейся в результате первичной клональной экспансии инфицирующего вирусного генома. Тесно связанные с ним варианты, не узнаваемые нейтрализующими антителами, обозначены как V2. Индукция последующих антительных ответов показана на нижних двух панелях. Антитела появляются вследствие индукции V3-специфических клонов В-клеток, которые продолжают экспансию из-за перекрестной реактивации близкими вариантами вируса, т.е. V2. По [43]
Рисунок 4. Модель феномена «первичного антигенного греха» при ВИЧ-инфекции у людей, вызванного презентацией иммунной системе gp120. Заштрихованная область, обозначенная как V1, соответствует вирусной нагрузке, образовавшейся в результате первичной клональной экспансии инфицирующего вирусного генома. Тесно связанные с ним варианты, не узнаваемые нейтрализующими антителами, обозначены как V2. Индукция последующих антительных ответов показана на нижних двух панелях. Антитела появляются вследствие индукции V3-специфических клонов В-клеток, которые продолжают экспансию из-за перекрестной реактивации близкими вариантами вируса, т.е. V2. По [43]

При изучении протективного эффекта ВИЧ-вакцины, включающей в качестве антигенного компонента gp120.16, выделенный из ВИЧ-1 SF2, получены сходные результаты. Люди, вакцинированные такой вакциной и имеющие высокие титры антител к gp120.16, оказались восприимчивы к вариантам ВИЧ-1, циркулирующим в их популяции. При развитии ВИЧ-инфекции в их сыворотке крови преобладали антитела к gp120.16 ВИЧ-1 SF2, а не к такому же оболочечному гликопротеину вируса, вызвавшего инфекцию [46].
Феномен антигенного импринтинга обнаружен и при изучении иммунного ответа у ВИЧ-инфицированных пациентов. Выработка антител к ВИЧ у них имеет олигоклональный характер. Одновременно происходит нарушение соотношения κ/λ типов легких цепей антител, поддерживающееся в течение многих лет независимо от скорости прогрессирования заболевания. Ограниченные (restricted) и при этом стабильно поддерживающиеся антительные ответы на антигены ВИЧ у таких пациентов представляют собой одну из причин невозможности выработки антител к ВИЧ-1, которые эффективно связывали бы сероварианты вируса, образовавшиеся в ходе персистирующего инфекционного процесса [47].
Антигенный импринтинг при малярии. Благодаря работам Pleass et al. [48] удалось показать возможность создания противомалярийной вакцины на основе 19-кДа фрагмента белка MSP119, находящегося на поверхности мерозоитов Plasmodium falciparum — бесполых форм плазмодия. При разрыве эритроцитов мерозоиты попадают в кровь, что приводит к периодическим приступам лихорадки. Связывание специфических антител с белком MSP119 блокирует проникновение возбудителя малярии в эритроциты и активирует его уничтожение фагоцитами.
Wipasa et al. [49] в опытах на мышах смоделировали ситуацию гетерогенного ответа на вакцинацию белком MSP119 в популяции людей, длительно живущих в эндемичном по малярии регионе. Ими показано, что заражение мышей P.yoelii YM[8] вызывает образование антител к нативному MSP119, титр которых после перенесенной мышами экспериментальной малярии они повысили бустерной вакцинацией рекомбинантным белком MSP119. Однако действие, выполненное в обратном порядке, т.е. сначала однократная инъекция (субоптимальная вакцинация) рекомбинантного белка MSP119, а затем инфицирование P.yoelii YM, привело к образованию антител к MSP119, не обладающих протективным действием, и к снижению естественного иммунитета к заражению возбудителем малярии.
Антигенный импринтинг при лихорадке Денге. Лихорадка Денге — трансмиссивная болезнь, встречающаяся в странах Южной и Юго-Восточной Азии, Африки, Океании и Карибского бассейна. Отдельные вспышки болезни охватывают сотни тысяч человек. Ежегодно в мире не менее 50 млн человек заболевают лихорадкой Денге. Возбудитель лихорадки Денге (Dengue fever virus, DENV) — оболочечный (+)ssРНК-вирус[9], четыре серотипа которого (DENV1-DENV4) относятся к арбовирусам семейства Togaviridae рода Flavivirus (арбовирусы антигенной группы В). Передача возбудителя инфекции среди людей осуществляется комарами Aedes aegypti, среди обезьян — Aedes albopictus. Обычно болезнь имеет мягкое течение и может проходить бессимптомно. В 1–5 % случаев она приобретает характер геморрагической лихорадки (hemorrhagic fever — DHF). У заболевшего человека развиваются геморрагический диатез и шоковое состояние (шоковый синдром Денге), которые могут привести его к смерти [50]. Причины такого осложнения длительное время не были ясны.
В 1983 г. Halstead et al. [51] обнаружили, что у тайских детей, попавших в клинику в шоковом состоянии после повторного развития у них лихорадки Денге, в сыворотке крови обнаруживаются в основном антитела, специфичные к вирусам серотипов, вызвавших лихорадку Денге несколько месяцев назад. К серотипам вирусов, обнаруженным у маленьких пациентов вирусологическими методами исследования, антитела образовывались медленно и присутствовали в сыворотке пациентов в низких титрах. Исследователи объяснили данный феномен стимуляцией В-клеток памяти, оставшихся после первого инфицирования, т.е. антигенным импринтингом.
Основными антигенами вируса Денге, в отношении которых плазмоцитами синтезируются нейтрализующие антитела, являются оболочечный белок Е и премембранный белок prM. Главную роль в антигенном импринтинге играют эпитопы третьего домена белка Е (ED3). В отношении их происходит выработка антител с широкой перекрестной активностью к белку Е вирусов Денге других серотипов, обладающих низкой авидностью [32][10]. Однако антигенный импринтинг оказался только частью патогенетического механизма развития DHF, в котором участвует иммунная система.
Образующиеся в ответ на повторное инфицирование вирусом другого серотипа антитела к вирусам серотипа, вызвавшего первый инфекционный процесс, обладают перекрестной специфичностью к штамму вируса, вызвавшего повторное инфицирование пациента, но они не нейтрализуют его, а способствуют размножению в организме человека, связывая вирусные частицы с Fc-рецепторами (FcR)[11] на поверхности макрофагов/моноцитов. Данный феномен называется антителозависимым усилением инфекции и подробно описан ниже.
Суть феномена антигенного импринтинга. Приведенные выше данные и обобщение Nara et al. [43] позволяют кратко изложить суть феномена антигенного импринтинга при инфекционных и поствакцинальных процессах. При повторном контакте иммунной системы с патогенным микроорганизмом или вакциной различия между старым вариантом эпитопа антигена и его новым вариантом могут быть «незамеченными» иммунной системой примерно так, как в оптический прибор не различаются детали, выходящие за пределы его разрешения. И тогда в процессе антигенной стимуляции первыми активизируются В-клетки памяти, «запомнившие» предыдущий антиген. Далее они дифференцируются в плазмоциты, продуцирующие антитела в отношении этого антигена, хотя иммунная система с ним не контактирует. Образующиеся антитела не способны эффективно нейтрализовать возбудитель инфекционной болезни, выработка же специфических к нему антител тормозится из-за подавления «наивных» В-клеток активизировавшимися В-клетками памяти. Как заметили Kim et al. [16], в данном случае В-клетки памяти формируют «слепое пятно» (blind spot) иммунной системы. Parsons et al. [42] такой ответ В-клеток памяти назвали замороженным репертуаром (repertoire freeze). Закон Дженнера — Пастера и правило Бернета соблюдаются, но при антигенной дистанции между штаммами (серотипами, серовариантами) возбудителя инфекционной болезни, превышающей размеры «слепого пятна» иммунной системы.
Для возбудителей инфекционных болезней, вызывающих феномен антигенного импринтинга, характерны:
-ограниченность антигенных эпитопов;
-большое количество карбонгидратов, экранирующих эпитопы, и/или ограниченные иммунодоминантные эпитопы;
-перекрестно-реактивные детерминанты у семейств малосвязанных патогенов;
-выраженная и олигомерная презентация эпитопов иммунной системе;
-незначительные различия в аминокислотных последовательностях или в форме антигена и гомологичных белков хозяина;
-анамнестический ответ, наступающий вслед за введением гетерологичного антигена;
-рекуррентная инфекция или бустинг (повторная иммунизация с целью усиления иммунного ответа) вирусами или антигенами, увеличивающие гуморальный иммунный ответ к первоначальному инфекционному агенту или антигену;
-пул длительно живущих клеток В-памяти;
-олигоклональный сывороточный профиль (т.е. происходит ответ только на доминантные эпитопы);
-преобладание клонально-производных В-клеток и популяций антител, специфичных для эпитопа.
Роль антигенного импринтинга в эпидемических, инфекционных и поствакцинальных процессах. В общем виде ее можно представить следующим образом:
1)антигенный импринтинг, развившийся в ответ на инфекционный процесс или вакцинацию, сопровождает человека всю его жизнь и предопределяет реакцию его иммунной системы в инфекционных процессах и структуру заболеваемости населения во время эпидемий (пандемий), вызванных тем же возбудителем инфекционной болезни;
2)при полном антигенном совпадении с возбудителем болезни, сформировавшим В-клетки памяти в некотором прошлом, этими клетками вырабатываются специфические антитела, обладающие протективным действием; иммунный ответ на возбудитель болезни носит выраженный протективный характер, и развития инфекционного процесса может не произойти. В результате ретроспективного эпидемиологического анализа будут обнаружены возрастные группы населения, оказавшиеся не вовлеченными в эпидемию (пандемию);
3)если между возбудителями инфекционной болезни, вызвавшими первый и второй инфекционные процессы, нет антигенного совпадения, но антигенная дистанция между ними настолько мала, что иммунная система не может отличить штамм возбудителя инфекционной болезни от того, что сформировали В-клетки памяти во время первого инфекционного процесса, то плазмоциты синтезируют антитела, специфичные к штамму возбудителя инфекционной болезни, распространявшегося в ту пандемию, когда сформировались В-клетки памяти. В результате иммунная система «отрабатывает» ложную цель, защитный эффект отсутствует. При ретроспективном эпидемиологическом анализе будут обнаружены возрастные группы населения, понесшие наибольшие потери в данную пандемию;
4)при проявлении антигеного импринтинга в ответ на возбудитель инфекционной болезни или вакцинацию, кроме антител, специфичных к антигену, распознанному иммунной системой человека первым, будут образовываться антитела, реагирующие перекрестно с возбудителями близких по антигенной структуре штаммов, но обладающие по отношению к ним низкой авидностью и способные усиливать инфекционный процесс (эффект антителозависимого усиления инфекции, см. ниже);
5)если антигенная дистанция между штаммом возбудителя инфекционной болезни, вызвавшим инфекционный процесс в прошлом, и штаммом, вызвавшим новый инфекционный процесс, настолько велика, что иммунная система его распознает, то иммунный ответ может быть направлен на противодействие этому штамму. Одновременно сформируются новые В-клетки памяти, которые при последующих вспышках этой же инфекционной болезни будут реагировать с возбудителем болезни, так как описано выше (п. 1–4);
6)при возможности развития феномена антигенного импринтинга многократная вакцинация и перенесенные инфекционные болезни делают малопредсказуемыми ответы иммунной системы на повторное заражение этими же возбудителями инфекционной болезни — от иммунитета, предотвращающего развитие инфекционной болезни, до ее утяжеления с летальными исходами у заболевших. Поствакцинальные осложнения, связанные с антигенным импринтингом, могут проявляться через десятилетия после ее проведения. Одна и та же вакцина может дать противоположные результаты в группах населения, имеющих разную эпидемическую историю и ранее многократно вакцинированных этой же вакциной.
Устранение антигенного импринтинга при вакцинации. В табл. 2 обобщены подходы к устранению антигенного импринтинга при вакцинации.
Таблица 2. Устранение антигенного импринтинга при вакцинации
|
Способ устранения антигенного импринтинга |
Возбудитель инфекционной болезни |
За счет чего устраняется антигенный импринтинг |
Источник |
|
Удаление перекрестно-реагирующих эпитопов у доминантного антигена |
Вирус Денге |
Авторы пошли по пути создания узко специфических моновакцин. Ими разработана ДНК-вакцина на основе плазмиды pVD1-CRR с клонированными структурными генами оболочечного белка Е и премембранного белка (prM). Замены аминокислотных последовательностей проведены в двух регионах белка Е, необходимых для синтеза перекрестно-реагирующих антител (домен II высоко консервативного белка слияния и A-цепи и DE-петли домена III). После поглощения ДНК-вакцины клетками реципиента, структурные гены антигенов транскрибируются и транслируются. Клетки синтезируют белки самособирающиеся в вирус-подобные частицы (virus-like particles, VLPs), презентирующие эпитопы белка Е Т- и В-лимфоцитам |
[31] |
|
Активизация дендритных клеток специальными адъювантами |
Вирус гриппа |
Мыши последовательно заражались близкими по гемагглютинину и нейраминидазе вирусами гриппа сероподтипа H1N1 (A/PR/8/34 и A/FM/1/47). Использование в качестве адъювантов токсина Bordetella pertussis (PT), CpG ODN (синтетические олигодезоксинуклеотиды, содержащие CpG-мотивы) или наноэмульсии, включающей сквален «масло в воде» (NE), позволяло устранить антигенный импринтинг, если их вводили при первой и второй вакцинациях |
[33] |
|
Стратегия гетерологичного примирующего бустинга (heterologous prime-boost strategy) |
ВИЧ |
Последовательная замена антигенов при бустерных и примирующих вакцинациях. Пример такой стратегии – исследование в Тайланде эффективности ВИЧ-вакцины. Первым вакцинным компонентом был рекомбинантный вектор на основе поксвируса канарейки (recombinant canary pox vector) ALVAC-HIV (vCP1521). Вектор экспрессировал гены оболочечных белков ВИЧ-1 (из CRF01_AE 92TH023 и LAI-вирусов), gag (LAI) и протеазу (LAI). Второй вакцинирующий компонент представлял собой смесь их двух CHO-производных оболочечных белков ВИЧ-1, сорбированных на алюминии, и названных AIDSVAX B/E. Первым вводили пациентам на 1 и 4 неделе vCP1521. Затем, через 12 и 24 нед. им вводили vCP1521, смешанный с AIDSVAX B/E |
[52] |
Судя по датам поступления в редакции научных журналов рукописей статей, работы по созданию нового поколения вакцин, позволяющих «обойти» антигенный импринтинг при массовых вакцинациях населения, ведутся уже не менее 10 лет. Основываются они на объективных знаниях тонких механизмов иммунного ответа применительно к инфекционному процессу, вызванному конкретным возбудителем инфекционной болезни.
3.Феномен антителозависимого усиления инфекции (antibody-dependent enhancement — ADE)[12]
Феномен ADE впервые описан Hawkes в 1964 г. [53], обнаружившим повышение продукции различных флавивирусов (японского энцефалита, энцефалита долины Мюррей и др.) в клетках куриного эмбриона, впервые экспонированных к вирусам, находящимся в среде с низким содержанием специфических антител. Впоследствии он привел доказательства, что увеличение «выхода» вируса в подобных экспериментах вызвано образованием комплекса «вирус — антитело» [54]. Эти данные настолько расходились с общепринятыми представлениями о защитной роли антител в инфекционном процессе, что их посчитали артефактами. Однако в конце 1960-х и начале 1970-х гг. уже другими исследователями обнаружена роль ADE в патогенезе тяжелых форм геморрагической лихорадки, вызванной вирусом Денге. Было установлено, что наличие антител в сыворотке крови реконвалесцента, оставшихся после легко перенесенных случаев лихорадки Денге, приводит к тяжелому течению болезни, если произошло повторное заражение DENV другого серотипа [55, 56]. За рубежом феномен ADE систематически изучается с конца 1980-х гг. [57] Но его описание в российских руководствах для врачей не приводилось.
Стадии ADE. Суть феномена ADE состоит в усилении инфекционного процесса в присутствии антител, специфических к возбудителю инфекционной болезни. ADE развивается в две стадии:
-внешнее ADE (extrinsic ADE — eADE) — вирус–специфическое антитело, образовавшее комплекс с вирусом посредством взаимодействия его Fc-фрагмента[13] с рецептором Fc (FcR)[14] и/или с рецепторами комплемента на поверхности фагоцитирующих клеток, усиливает распространение вируса по фагоцитирующим клеткам;
-внутреннее ADE (intrinsic ADE, iADE) — комплексы «вирус — специфическое антитело»[15], взаимодействующие с фагоцитирующей клеткой через Fc-рецепторы и рецепторы комплемента, запускают сигнальные механизмы, блокирующие ее антивирусную защиту, и тем самым способствуют внутриклеточному размножению вируса.
В основном феномен ADE проявляется в ответ на образование антител изотипа IgG1 [58]. У людей имеются три типа рецепторов Fc, которые связывают IgG: сиалогликопротеины FcyRI, FcyRII и FcyRIII (CD16). FcyRI наиболее представлен на моноцитах/макрофагах человека, и он связывает IgG с наибольшей авидностью. Поэтому ему принадлежит лидерство среди других рецепторов макрофагов в реализации феномена ADE. ADE, показанный в условиях in vitro, не обязательно воспроизводится в условиях in vivo [59].
Феномен ADE характерен для инфекционных процессов, вызываемых вирусами, имеющими следующие особенности:
а)обычно они реплицируются в макрофагах;
б)индуцируют продукцию большого количества антител с низкой способностью к нейтрализации гомологичных вирусов;
в)способны к персистентной инфекции, характеризующейся продолжительной виремией [60].
Феномен ADE также обнаружен при инфекционных процессах, вызываемых бактериальными патогенами, но изучен фрагментарно. Например, порообразующий токсин золотистого стафилококка — лейкоцидин усиливает свое токсическое действие, если в крови человека содержатся специфические к нему антитела [61]. Такой же эффект вызывают моноклональные антитела к токсину А патогенных клостридий [62]. Имеются косвенные доказательства причастности феномена ADE к прогрессированию туберкулезной инфекции и Ку-лихорадки. При аэрозольном инфицировании M.tuberculosis мышей 57BL/6, дефицитных по рецептору FcgIIB, патологические изменения у них развиваются через 30 сут., у интактных мышей — через 20 сут. [63]. В условиях in vitro показано, что антитела к C.burnetii I фазы стимулируют ее размножение в макрофагах более эффективно, чем антитела к этому же микроорганизму II фазы [64, 65].
Возможно, что первое описание ADE дал Заболотный, наблюдавший в 1899 г. в Вэнчане (Монголия) появление пустулезной формы чумы у больного с бубонной чумой на пятые сутки после введения противочумной сыворотки. Он объяснил это явление примерно так, как сегодня объясняют ADE: антитела к возбудителю чумы распространили его по фагоцитирующим клеткам и усилили инфекционный процесс [67]. Можно предположить, что из-за низкого качества противочумной сыворотки, примененной Д.К. Заболотным, и ненадлежащих условий ее хранения во время экспедиции к очагам чумы в Монголии антитела к возбудителю чумы утратили нейтрализующее действие, но сохранили способность взаимодействовать с FcR.
eADE. Феномен наблюдается в двух вариантах:
а)комплемент-опосредованное антителозависимое усиление инфекции (complement-mediated ADE — C-ADE);
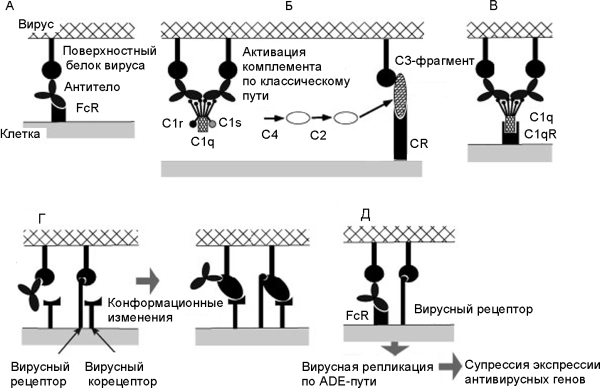
б) независящее от комплемента и связанное с Fc-рецептором усиление инфекции (Fc-receptor-mediated ADE — FcR-ADE) [60, 68] (рис. 5).
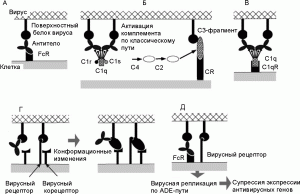 Рисунок 5. Общая схема развития феномена eADE при вирусных инфекциях. А. Взаимодействие между антителом и FcR на поверхности макрофага. Б. Фрагмент С3 комплемента (компонент комплемента, после присоединения которого весь этот комплекс приобретает способность прилипать к различным частицам и клеткам) и рецептор комплемента (complement receptor; CR) способствуют присоединению вируса к клетке. В. Белки комплемента С1q и С1qR способствуют присоединению вируса к клетке (в составе молекулы C1q имеется рецептор для связывания с Fc-фрагментом молекулы антитела). Г. Антитела взаимодействуют с рецептор-связывающим сайтом вирусного белка и индуцируют его конформационные изменения, облегчающие слияние вируса с мембраной. Д. Вирусы, получившие возможность реплицироваться в данной клетке посредством eADE, супрессируют антивирусные ответы со стороны антивирусных генов клетки. По [68]
Рисунок 5. Общая схема развития феномена eADE при вирусных инфекциях. А. Взаимодействие между антителом и FcR на поверхности макрофага. Б. Фрагмент С3 комплемента (компонент комплемента, после присоединения которого весь этот комплекс приобретает способность прилипать к различным частицам и клеткам) и рецептор комплемента (complement receptor; CR) способствуют присоединению вируса к клетке. В. Белки комплемента С1q и С1qR способствуют присоединению вируса к клетке (в составе молекулы C1q имеется рецептор для связывания с Fc-фрагментом молекулы антитела). Г. Антитела взаимодействуют с рецептор-связывающим сайтом вирусного белка и индуцируют его конформационные изменения, облегчающие слияние вируса с мембраной. Д. Вирусы, получившие возможность реплицироваться в данной клетке посредством eADE, супрессируют антивирусные ответы со стороны антивирусных генов клетки. По [68]
В табл. 3 обобщены сведения по вирусным и бактериальным инфекциям, сопровождающимся феноменом еADE.
Таблица 3. Инфекционные болезни, сопровождающиеся феноменом еADE*
|
Инфекционная болезнь |
Возбудитель болезни (семейство) |
Тип ADE |
Примечание |
Источник |
|
Вирусные инфекции |
||||
|
ВИЧ/СПИД |
Human immunodeficiency virus, HIV, ВИЧ (Retroviridae) |
FcR-ADE, C-ADE, |
На ранней стадии инфекции феномен реализуется через V3-петлю gp120 (по типу FcR-ADE). По типу C-ADE феномен начинает проявляться перед клиническим прогрессированием ВИЧ-инфекции |
[57, 69, 70, 75] |
|
Инфекционная анемия лошадей |
Equine infectious anemia virus, EIAV (Retroviridae) |
FcR-ADE |
Показано обострение болезни у вакцинированных лошадей и пони, как следствие присутствия антител, индуцированных введением инактивированной цельновирионной вакцины или рекомбинантной вакцины, полученной на основе поверхностного гликопротеина EIAV |
[71, 72]
|
|
Иммунодефицит кошек |
Feline immunodeficiency virus, FIV (Retroviridae) |
FcR-ADE |
Вакцинирование оболочечным рекомбинантным белком вируса усиливает виремию. Усиление клинических признаков болезни у кошек при росте титров антител к коровому белку (core protein) FIV |
[73]
|
|
Инфекционный перитонит кошек |
Feline infectious peritonitis virus, FIPV, (Coronaviridae) |
FcR-ADE |
Феномен ADE проявляется при инфицировании вирусом того же серотипа, в отношении которого был иммунный ответ |
[74] |
|
Лихорадка Эбола |
Вирус Эбола (Filoviridae) |
FcR-ADE, C-ADE |
Сыворотка, взятая от мышей, иммунизированных ДНК вакциной с геном поверхностного гликопротеина вируса Zaire, вызывает ADE |
[76] |
|
Лихорадка Марбург |
Вирус Марбург (Filoviridae) |
C-ADE |
ADE наиболее выражен при инфекции, вызванной вирулентный изолятом Angola |
[77] |
|
Гепатит С |
Вирус гепатита С (Flaviviridae) |
FcR-ADE |
ADE рассматривается как основная причина, мешающая созданию эффективных вакцин |
[78] |
|
Желтая лихорадка |
Viscerophilus tropicus (Flaviviridae) |
FcR-ADE |
ADE рассматривается как основная причина нейровирулентности вируса и тяжелого течения болезни. Показана связь ADE с антителами к гликопротеину Е вируса |
[79, 80]
|
|
Корь |
Measles virus (Paramyxoviridae) |
FcR-ADE |
ADE вызывают антитела к гемагглютинину Н. Авторы связывают с ADE случаи так называемой атипичной кори, когда она развивается у ранее вакцинированных людей и протекает в тяжелой форме |
[81] |
|
Энцефалит Западного Нила |
West Nile virus, WNV (Flaviviridae) |
C-ADE |
ADE вызывают субклассы IgG, с высокой афинностью к фактору комплемента C1q |
[82] |
|
Энцефалит долины Мюррей |
Murray Valley encephalitis virus, MVEV (Flaviviridae) |
Нет данных |
Антитела к вирусу японского энцефалита в субнейтрализующих концентрациях увеличивают вирусемию и смертность среди мышей, зараженных MVEV. Авторы выражают опасение, что программы по вакцинации населения к вирусу японского энцефалита в тех районах, где одновременно с ним циркулирует и MVEV, могут способствовать развитию эпидемии энцефалита долины Мюррей |
[83] |
|
Клещевой энцефалит |
Tick-borne encephalitis virus, TBEV (Flaviviridae) |
Нет данных |
Феномен показан в условиях in vitro. Высказано предположение о возможности развития ADE у людей на фоне низких титров специфических к вирусу антител |
[125] |
|
Лихорадка Денге |
Dengue virus, DENV, DV (Togaviridae) |
FcR-ADE |
Геморрагическая лихорадка развивается при перекрестном инфицировании любым из серотипов вируса |
[84]
|
|
Энтероинфекции и патология ЦНС |
Human enterovirus 71, EV71 (Picornaviridae) |
FcR-ADE |
Корреляция между ADE и тяжелым течением болезни |
[85] |
|
Бешенство |
Вирус бешенства (Rhabdoviridae) |
Нет данных |
Антитела к вирусу усиливают инфицирование макрофагов в условиях in vitro через образование иммунных комплексов. Предполагается связь этого феномена с «ранней смертью» от бешенства у вакцинированных животных |
[86, 87, 88] |
|
Алеутская болезнь норок |
Aleutian disease virus, ADV (Parvoviridae) |
FcR-ADE |
Комплекс «ADV–антитело» осаждается на ренальных гломерулярных мембранах или стенках капиллярных сосудов почек, вызывая летальный гломерулонефрит |
[89]
|
|
Бактериальные инфекции |
||||
|
Стафилококковая инфекция |
Лейкоцидин Пантон-Валентина (Panton-Valentine leukocidin, PVL) – пороформирующий цитотоксин золотистого стафилококка (Micrococcaceae) |
Нет данных |
Антитела к PVL, присутствующие в крови большинства людей, усиливают действие PVL, повышая патогенность PVL-продуцирующих S. aureus |
[61] |
|
Клостридиоз |
Токсин A, toxin TcdA Clostridium difficile (Clostridiaceae) |
FcR-ADE |
Моноклональные антитела к токсину А усиливают его поражающее действие |
[62] |
|
Туберкулез |
M. tuberculosis (Mycobacterium tuberculosis complex) |
FcR-ADE (предпо-ложи-тельно) |
У мышей 57BL/6, дефицитных по рецептору FcγIIB, при аэрозольном инфицировании M. tuberculosis патологические изменения развиваются медленнее, чем у интактных мышей |
[63] |
|
Лихорадка Ку |
Coxiella burnetii (Rickettsiella) |
Нет данных |
Антитела к C. burnetii I фазы стимулируют ее размножение в макрофагах |
[64]
|
* За основу взята таблица, опубликованная нами ранее [9, 10].
Безоболочечным вирусам (non-enveloped viruses), образовавшим комплекс с антителом, способным взаимодействовать с Fc-рецептором, специфические рецепторы на поверхности клетки-мишени не требуются [68].
Компонент комплемента С1[16], связывая Fc-фрагмент антитела, инициирует классический путь активации комплемента, в результате чего активируется компонент комплемента С3, ковалентно (!) связывающийся или с антителом, или с поверхностью вирусной частицы. Образовавшийся комплекс способен взаимодействовать с рецепторами комплемента на поверхности клетки посредством С3, усиливая взаимодействие вируса с клеткой. Альтернативно C1q-субъединица непосредственно может перекрестно связывать вирусный белок и C1q-рецепторы (C1qR) на поверхности фагоцитирующих клеток. Все перечисленные эффекты находятся в зависимости от концентрации антител.
iADE. Толчком к исследованиям блокирующего действия ADE на антивирусную защиту клетки послужили данные, полученные при изучении причин развития хронических артритов у реконвалесцентов, перенесших острую форму болезни, вызванную вирусом Росс Ривер (Ross River virus, RRV). Такие артриты могут длиться до года, делая пациента на весь этот период неработоспособным. В синовиальной жидкости пациентов с хроническими артритами обнаружены антигены RRV и гамма-интерферон (IFN-гамма), что свидетельствует о хронической RRV-инфекции. При попытке ее воспроизвести на линиях мышиных макрофагов и первичных человеческих моноцитов/макрофагов (primary human monocytes/macrophages)[17] установлено, что инкубирование RRV с разбавленной специфической сывороткой приводит:
1)к супрессии синтазы оксида азота 2 (nitric oxide synthase 2 — NOS2) и, соответственно, к снижению продукции активных радикалов азота (reactive nitrogen radical);
2)прекращению экспрессии генов интерферон-регулирующего фактора 1 (interferon regulatory factor 1, IRF-1) и фактора ядра каппа-би (nuclear factor kВ) и, соответственно, к подавлению синтеза фактора некроза опухолей альфа (tumor necrosis factor alpha — TNF-alpha) и IFN-γ;
3) заметному увеличению синтеза интерлейкина-10 (interleukin-10 — IL-10). Лигирование FcyR с комплексом «антитело — зимозан» в присутствии RRV не вызывало вышеописанного эффекта [90, 91]. Следовательно, увеличение продукции вируса клетками при ADE вызвано не только увеличением возможностей для взаимодействия вируса с поверхностью макрофагов, но и подавлением их собственной системы защиты от вирусов (innate cellular immunity) [101].
Классификация феномена ADE. Приведенные данные позволяют нам предложить классификацию феноменов ADE по двум принципам: по типу рецептора, с которым вирус взаимодействует на поверхности моноцитов/макрофагов (С-ADE и FcR-ADE), и по механизмам развития ADE (рис. 6).
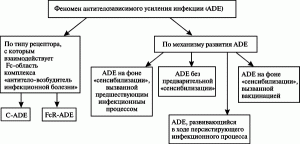 Рисунок 6. Классификация ADE. По [9]
Рисунок 6. Классификация ADE. По [9]
Первая классификация удобна для изучения феномена ADE в условиях in vitro, например для установления границ феномена среди близкородственных видов вирусов на клетках культур тканей, содержащих либо, наоборот, не содержащих Fc- и CIq-рецепторы, либо при их блокировании специфическими мАТ. Границы феномена ADE устанавливаются с помощью специфических сывороток к вирусам близкородственных видов. Вторая классификация — для воспроизведения ADE в условиях in vivo при разработке ИЛП и их доклинических и клинических исследованиях.
Феномен ADE, развивающийся на фоне сенсибилизации, вызванной предшествующим инфекционным процессом
Наиболее изучен среди других проявлений феномен ADE, поэтому мы рассмотрим его более подробно, чем остальные. Опережающим объектом исследований при изучении данного феномена является геморрагическая лихорадка Денге. Схема жизненного цикла DENV в отсутствие специфических антител представлена на рис. 7.
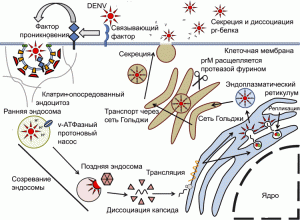 Рисунок 7. Жизненный цикл DENV в отсутствие специфических антител. Вирус проникает в клетку по механизму рецептор-опосредованного эндоцитоза. Клеточная мембрана впячивается внутрь клетки, формируя окаймленные ямки. Внутриклеточная сторона окаймленной ямки в основном включает белок клатрин (clathrin). В зависимости от серотипа вируса или типа клетки, вирус при рН 6,0 сливается со стенкой ранней эндосомы и покидает ее, либо это происходит при рН 5,0–6,0 уже в поздней эндосоме. В эндоплазматическом ретикулуме (endoplasmic reticulum, ER) (+)РНК транслируется на рибосомах с образованием отдельного полипротеина, который в последующем процессируется с помощью аутопротеаз и клеточных протеаз на 7 неструктурных белков (NS1, 2A, 2B, 3, 4A, 4B и 5) и три структурных белка: C (капсид), prM (прекурсорный мембранный белок, precursor membrane protein) и E-белок. Неструктурные белки инициируют репликацию РНК вируса, prM и E-белки формируют гетеродипмеры, которые ориентированы в просвет эндоплазматического ретикулума, где происходит частичная сборка вирионов. РНК вируса ассоциируется с С-белком и формирует нуклеокапсид, который «одевается» липидной мембраной, содержащей гетеродимеры prM- и Е-белков. Из цитоплазмы клетки вирионы выводятся через сеть Гольджи, где происходит их созревание – prM расщепляется сериновой протеазой фурином с образованием растворимого pr-белка и М-белка. pr-белок остается ассоциированным с Е-белком во время экзоцитоза, предотвращая преждевременное слияние вирионов с участками сети Гольджи, имеющими кислые значения рН. Попав в нейтральную среду межклеточного пространства pr-белок диссоциирует, и вирусная частица становится способной вызывать инфекционный процесс, т.е. созревшей. По [50]
Рисунок 7. Жизненный цикл DENV в отсутствие специфических антител. Вирус проникает в клетку по механизму рецептор-опосредованного эндоцитоза. Клеточная мембрана впячивается внутрь клетки, формируя окаймленные ямки. Внутриклеточная сторона окаймленной ямки в основном включает белок клатрин (clathrin). В зависимости от серотипа вируса или типа клетки, вирус при рН 6,0 сливается со стенкой ранней эндосомы и покидает ее, либо это происходит при рН 5,0–6,0 уже в поздней эндосоме. В эндоплазматическом ретикулуме (endoplasmic reticulum, ER) (+)РНК транслируется на рибосомах с образованием отдельного полипротеина, который в последующем процессируется с помощью аутопротеаз и клеточных протеаз на 7 неструктурных белков (NS1, 2A, 2B, 3, 4A, 4B и 5) и три структурных белка: C (капсид), prM (прекурсорный мембранный белок, precursor membrane protein) и E-белок. Неструктурные белки инициируют репликацию РНК вируса, prM и E-белки формируют гетеродипмеры, которые ориентированы в просвет эндоплазматического ретикулума, где происходит частичная сборка вирионов. РНК вируса ассоциируется с С-белком и формирует нуклеокапсид, который «одевается» липидной мембраной, содержащей гетеродимеры prM- и Е-белков. Из цитоплазмы клетки вирионы выводятся через сеть Гольджи, где происходит их созревание – prM расщепляется сериновой протеазой фурином с образованием растворимого pr-белка и М-белка. pr-белок остается ассоциированным с Е-белком во время экзоцитоза, предотвращая преждевременное слияние вирионов с участками сети Гольджи, имеющими кислые значения рН. Попав в нейтральную среду межклеточного пространства pr-белок диссоциирует, и вирусная частица становится способной вызывать инфекционный процесс, т.е. созревшей. По [50]
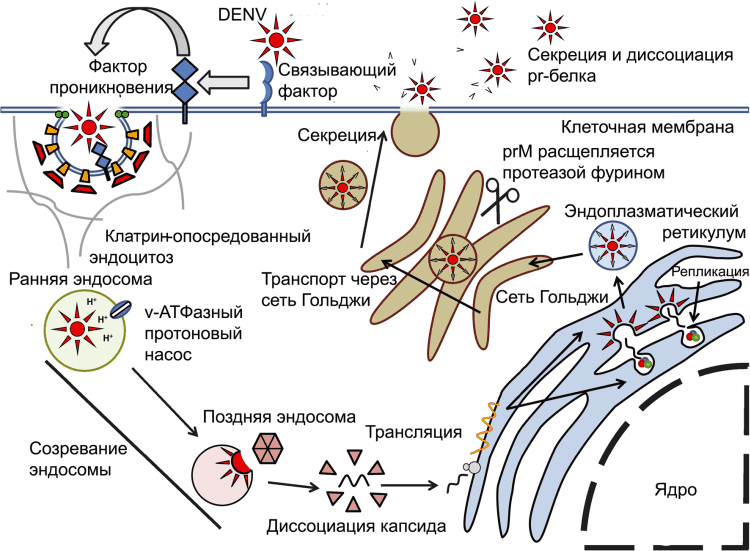
После проникновения DENV в эндосомы, клетка запускает механизмы антивирусной защиты [93], в частности экспрессию интерферонов (IFN). Оба типа интерферонов — тип I (a, b) и тип II (y) способны блокировать репликацию DENV, если происходит его распознавание эндосомальными рецепторами: toll-подобный рецептор 3 (toll-like receptor — TLR-3)[18] — распознает двухцепочечную РНК (dsRNA) вируса; TLR8 — распознает G-богатые олигонуклеотиды и TLR7 — распознает ssРНК.
В цитоплазме вирусную РНК «узнают» цитоплазматические РНК-геликазы (cytoplasmic RNA helicases)[19], RIGI (retinoic-acid inducible gene 1) и MDA5 (melanoma differentiation-associated gene 5). Активация TLR индуцирует экспрессию провоспалительных цитокинов: IL-8, IL-12, IFN-a и IFN-y. Положительная регуляция экспрессии IL-8 осуществляется через ядерный фактор каппа-би (NF-kB). Экспрессия IFN активирует STAT1 и усиливает экспрессию IRF1 (IFN regulatory factor 1), что приводит к усиленной продукции активных радикалов азота (NO). Комбинированное действие интерферонов и NO вызывает антивирусное состояние у соседних клеток (antiviral state) и ограничивает размножение DENV в инфицированных клетках соответственно [94].
При первичном инфицировании человека DENV иммунные ответы на вирус мало отличаются от тех, что описаны в классической схеме иммунного ответа, приведенной выше. Специфичные в отношении DENV B- и T-клетки формируются приблизительно через 6 сут. после инфицирования и полностью контролируют развитие инфекции. Вирион DENV распознается антителами, специфичными к белкам E и prM. Структурная организация этих белков у «созревшего» и «несозревшего» вируса различается. Следовательно, различаются и их специфические эпитопы. Доминирующую роль в нейтрализации вируса играют антитела к белку prM «созревшего» вируса. Нейтрализующая активность специфических к DENV антител проявляется на двух уровнях:
1)блокирование взаимодействия вируса с клеточным рецептором;
2)блокирование слияния вируса с клеточной мембраной вследствие связывания антителами петли слияния белка Е.
Антитела к prM «несозревшего» вируса обладают перекрестной активностью к DENV всех серотипов, но их нейтрализующая активность незначительна [95, 96].
Репликация DENV, как и любого другого РНК-вируса, сопровождается большим количеством ошибок. Вызвано это тем, что все молекулы вирусной РНК реплицируют через асимметричную транскрипцию с одной цепи, исключающую большинство корректирующих механизмов, характерных для репликации ДНК. Поэтому первичный инфекционный процесс при лихорадке Денге сопровождается полиморфизацией DENV и образованием квазиспецифичных производных в пределах его серотипа. Иммунная система реагирует на них выработкой специфических антител [97].
При вторичном инфицировании человека DENV гетерологичного серотипа стимулируются клоны В-клеток памяти, сохраняющие информацию о DENV, инфицировавшем человека первично. Они дифференцируются в плазмоциты, продуцирующие антитела к вирусу (его квазипроизводным), который они запомнили, а не к тому, который вызвал инфекцию. Этот иммунологический феномен называется феноменом «первичного антигенного греха», или антигенным импринтингом (см. выше).
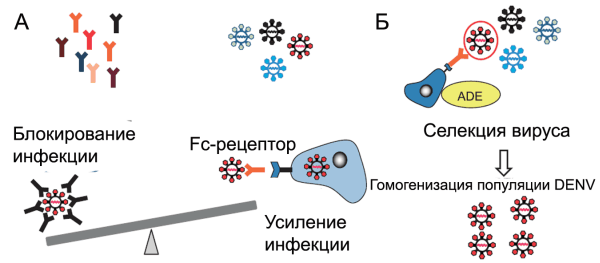
Усиление инфекционного процесса происходит еще до того, как концентрация антител достигнет порога, необходимого для нейтрализации вируса. Продуцируемые плазмоцитами антитела «узнают» DENV, вызвавший инфекционный процесс, но не нейтрализуют его. Они формируют с вирусом комплекс и связывают его с Fc-рецептором на поверхности макрофагов (феномен FcR-ADE), тем самым усиливая инфекционный процесс. Одновременно происходит гомогенизация популяции DENV, так как на этапе eADE преимущества в инфицировании макрофагов/моноцитов получают лишь те квазипроизводные DENV, в отношении которых плазмоцитами вырабатываются антитела, способные связать их с Fc-рецепторами [50, 97][20] (рис. 8).
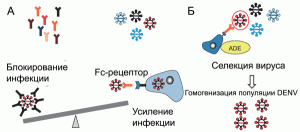 Рисунок 8. Механизм eADE при геморрагической лихорадке Денге. А. Вторичное инфицирование пациента DENV. На различных стадиях инфекции происходит дивергенция DENV. Уже существующие специфические антитела могут блокировать инфекционный процесс, если они встретились с тем его серотипом, который вызвал первичную инфекцию, или, наоборот, усиливать его через механизм ADE, если возбудитель болезни представлен вирусом другого серотипа. Б. Гипотетический механизм гомогенизации популяции DENV на этапе eADE. По [97]
Рисунок 8. Механизм eADE при геморрагической лихорадке Денге. А. Вторичное инфицирование пациента DENV. На различных стадиях инфекции происходит дивергенция DENV. Уже существующие специфические антитела могут блокировать инфекционный процесс, если они встретились с тем его серотипом, который вызвал первичную инфекцию, или, наоборот, усиливать его через механизм ADE, если возбудитель болезни представлен вирусом другого серотипа. Б. Гипотетический механизм гомогенизации популяции DENV на этапе eADE. По [97]
Изменения в клетке, связанные с iADE, начинаются раньше, чем DENV покинет эндосому. Точный механизм развития iADE не установлен. Имеющиеся знания позволили [50] описать его следующим образом.
Комплекс «DENV — специфическое антитело» через рецептор Fc запускает негативные регуляторы экспрессии TLR3, TLR4, TLR7 и TLR–сигнальных молекул. В результате слабой экспрессии этих рецепторов вирус, проникший в эндосому, не узнается клеткой, эффективной экспрессии генов, кодирующих интерфероны и синтез противовоспалительных цитокинов IL–8, IL–12, не происходит. Одновременно блокируется экспрессия IRF1, что тормозит продукцию активных радикалов азота[21]. Подавление системы противовирусной защиты клетки приводит к длительному размножению в них DENV и увеличению выхода зрелых вирусных частиц [97]. Однако только персистированием DENV в макрофагах iADE при лихорадке Денге не ограничивается.
По сигнальным путям, инициируемым через рецептор Fc, запускается экспрессия гена IL-10, макрофаг начинает продуцировать большие количества IL–10, ингибирующего синтез противовоспалительных цитокинов (IFN-y, IL-2, -3, -12 и др.) и усиливающего синтез фактора некроза опухолей (TNF) и IL-6, вызывающих повышенную проницаемость сосудов [99]. IL-10 также нарушает дифференциацию Т-хелперов на субпопуляции Th1 и Th2, что ведет к нарушению взаимодействия между клеточными и гуморальными звеньями иммунной системы при блокировании размножения DENV [100]. Лихорадка Денге развивается в тяжелой клинической форме.
Исследования, проведенные с целью выяснить, какие аминокислотные замены структурных и неструктурных белков различных серотипов DENV (мутации в их генах) ассоциируются с тяжелым течением болезни, не дали результатов. Повышенная виремия и высокие количества IL-10 в сыворотке крови всегда сопровождают тяжелое состояние больного. Других объяснений тяжелых осложнений при геморрагической лихорадке Денге, кроме как вовлечения в патогенез болезни ADE, пока не предложено [97, 101].
Феномен ADE, развивающийся без предварительной сенсибилизациииммунной системы. Takada et al. [68, 76, 102] показали, что ADE при инфекционном процессе, вызванном вирусом Эбола (субтип Zaire), развивается в результате взаимодействия образующихся вирус-специфических антител с вирусом и Fc1-рецептором или компонентом комплемента C1q и его рецептором (C1ADE) у макрофагов. Используя моноклональные антитела, исследователи локализовали такие эпитопы у GP вируса субтипа Zaire и сконструировали химерные эпитопы, индуцирующие продукцию антител у мышей со сниженной способностью вызывать ADE, но обладающих нейтрализующей активностью в отношении вируса субтипа Zaire. Феномен ADE был менее выражен для не опасного для человека субтипа Reston, чем для вирусов субтипов Zaire и Sudan. Авторы данных работ предположили, что феномен ADE играет важную роль в патогенезе лихорадки Эбола (рис. 9).
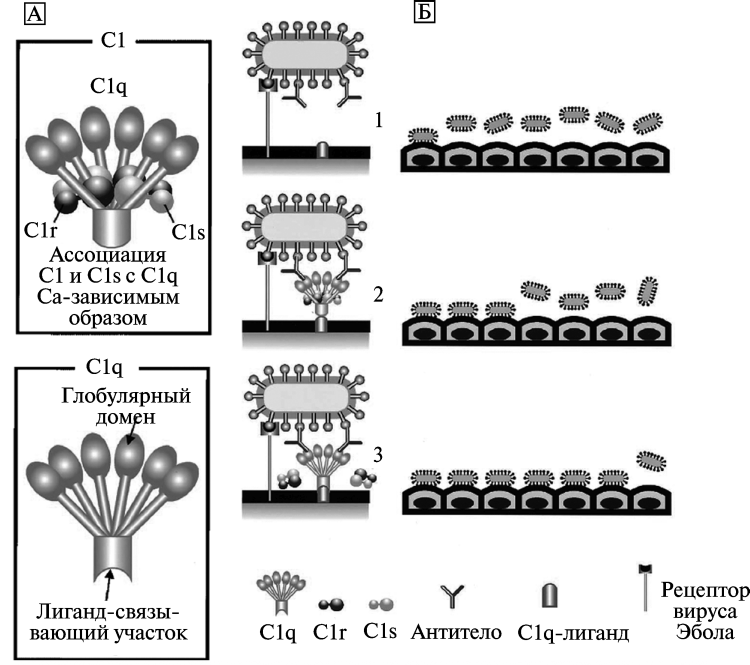
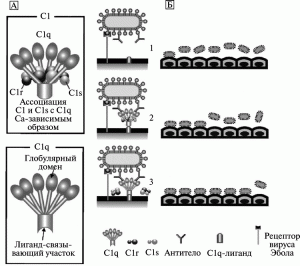 Рисунок 9. Модель C-ADE (C1q-ADE) при лихорадке Эбола. A. Схематическое изображение белков комплемента C1 и C1q. Молекула C1q включает глобулярный и лигандсвязывающий домены. Глобулярный домен состоит из шести глобулярных набалдашников (globular heads), которые связываются с Fc1-участком антитела. Лигандсвязывающий домен C1q взаимодействует с лигандом на поверхности фагоцитирующей клетки. Аффинитет C1q к лиганду снижается при ассоциации с C1r и C1s (сериновые протеазы). Б. Механизм ADE при лихорадке Эбола. Вирус Эбола инициирует инфекционный процесс путем связывания со специфическими рецепторами на поверхности фагоцитирующей клетки (1). C1q связывает комплекс «вирус–антитело» с C1q1-лигандами, расположенными на поверхности клеток, вызывая взаимодействие между вирусом и рецептором (2). Диссоциация C1r и C1s от C1q увеличивает связывающий аффинитет молекулы C1q с лигандами на поверхности фагоцитирующей клетки (3). По [68]
Рисунок 9. Модель C-ADE (C1q-ADE) при лихорадке Эбола. A. Схематическое изображение белков комплемента C1 и C1q. Молекула C1q включает глобулярный и лигандсвязывающий домены. Глобулярный домен состоит из шести глобулярных набалдашников (globular heads), которые связываются с Fc1-участком антитела. Лигандсвязывающий домен C1q взаимодействует с лигандом на поверхности фагоцитирующей клетки. Аффинитет C1q к лиганду снижается при ассоциации с C1r и C1s (сериновые протеазы). Б. Механизм ADE при лихорадке Эбола. Вирус Эбола инициирует инфекционный процесс путем связывания со специфическими рецепторами на поверхности фагоцитирующей клетки (1). C1q связывает комплекс «вирус–антитело» с C1q1-лигандами, расположенными на поверхности клеток, вызывая взаимодействие между вирусом и рецептором (2). Диссоциация C1r и C1s от C1q увеличивает связывающий аффинитет молекулы C1q с лигандами на поверхности фагоцитирующей клетки (3). По [68]
Для лихорадки Марбург феномен ADE был описан в 2011 г. Так же, как для субтипов вируса Эбола, показана связь между ADE и вирулентностью изолятов вируса Марбург. Авторами делается вывод, что феномен ADE лежит в основе патогенеза не только лихорадок Марбург и Эбола, но и других филовирусных геморрагических лихорадок [77].
Феномен ADE, развивающийся в ходе персистирующего инфекционного процесса. Феномен ADE лежит в основе патогенеза болезни многих персистирующих инфекционных процессов. Например, клинически выраженный кошачий инфекционный перитонит, вызываемый FIPV (семейство Coronaviridae), развивается у кошек, уже имевших антитела после ранее перенесенной бессимптомной инфекции либо на фоне персистирующей инфекции в случае мутации вируса, приведшей к появлению его нового антигенного варианта. Отличить же вирулентные штаммы FIPV от невирулентных в прямых опытах на животных не удается [103, 104].
Алеутская болезнь норок вызывается парвовирусом (Aleutian disease virus, ADV) из семейства Parvoviridae. ADV патогенен для норок всех цветных вариантов. Основной источник вируса — переболевшие норки-вирусоносители, выделяющие вирус с мочой, калом и слюной. Репликация ADV в макрофагах сопровождается секрецией плазматическими клетками большого количества антител, не обладающих способностью нейтрализовать вирус. Эти антитела образуют иммунные комплексы с ADV, увеличивающие инфицированность макрофагов и вызывающие образование ненейтрализующих антител. Порочный круг замыкается осаждением комплекса «ADV — антитело» на ренальных гломерулярных мембранах или стенках капиллярных сосудов почек, что приводит к летальному гломерулонефриту [89].
Но наиболее интересную роль феномен ADE играет при ВИЧ-инфекции. Для ВИЧ он показан в конце 1980-х гг. [69, 70], но до сих пор игнорируется разработчиками ВИЧ-вакцин.
У ВИЧ-инфицированных людей соблюдается определенная очередность проявления вариантов развития eADE. На ранней стадии инфекции феномен реализуется через V3-петлю gp120 (по типу FcR-ADE); по типу C-ADE феномен начинает проявляться перед клиническим прогрессированием ВИЧ-инфекции [57]. Клиническое значение феномена ADE для ВИЧ — это прогрессирование инфекции и облегчение переноса вируса от матери к плоду [105]. Вне контекста представлений о роли ретровирусов в эволюции клеточных форм жизни и роли ADE в эволюции ВИЧ процесс накопления разных вариантов ВИЧ в популяциях людей выглядит случайным, как проявление некой способности ВИЧ «постоянно меняться». Но случайностей в этом процессе нет.
По данным Takeda et al. [106], в условиях in vitro добавление к клеткам моноцитов сыворотки ВИЧ-инфицированных людей в субнейтрализующих концентрациях значительно усиливает репликацию вируса, т.е. на ранних этапах выработки антител к новому серотипу вируса основную роль в усилении инфекционного процесса играет феномен ADE. Высокие концентрации такой сыворотки в условиях in vitro показывают вирус-нейтрализующую активность. Следовательно, ВИЧ не удается «увильнуть» от специфических антител, однако блокирования инфекционного процесса специфическими антителами в условиях in vivo не происходит. Высокая скорость мутаций при обратной транскрипции и высокая скорость репликации ВИЧ генерируют большое количество серовариантов ВИЧ. Особенно этот процесс дает о себе знать после сероконверсии и перехода болезни в асимптоматическую стадию.
Как только уровень антител, нейтрализующих данный серотип ВИЧ, достигает определенного порога, селекционируется вариант вируса, избегающий их нейтрализующего действия [107]. Выработка антител к нему начинается заново. И вновь путем вовлечения в инфекционный процесс феномена ADE новому серотипу вируса обеспечивается распространение по клеткам, содержащим на своей поверхности Fc–рецептор (ранняя стадия инфекции) и рецептор комплемента (перед клиническим прогрессированием ВИЧ-инфекции). С каждым новым серовариантом вируса цикл повторяется. Скорость появления как ВИЧ-нейтрализующих антител, так и избегающих их вирусов варьирует у разных лиц, однако сам цикл многократно повторяется на протяжении жизни ВИЧ-инфицированного человека и больного СПИДом [108], приводя к росту генетического разнообразия ВИЧ. Только по мере истощения иммунной системы и, соответственно, работы маховика ADE гетерогенизация ВИЧ прекращается. Эту закономерность хорошо иллюстрируют данные Shankarappa et al. [109].
У ВИЧ-инфицированных пациентов, так называемых умеренных прогрессоров (moderate progressors), в пределах асимптоматической стадии ВИЧ-инфекции Shankarappa et al. [109] выделяют три фазы дивергенции и три фазы роста разнообразия ВИЧ. Под дивергенцией (divergence) эти авторы понимают различия между нуклеотидной последовательностью исходного вируса и последовательностью вируса, полученного от ВИЧ-инфицированного человека через какое-то время после инфицирования. Под разнообразием (diversity) подразумеваются различия в нуклеотидных последовательностях ВИЧ в данной временной точке (рис. 10).
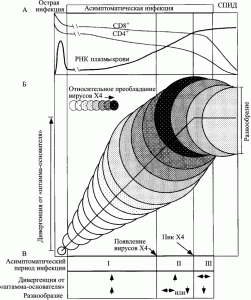 Рисунок 10. Схематическое изображение развития ВИЧ-инфекции у умеренных прогрессоров. Диаметры кругов приблизительно соответствуют разнообразию (diversity) вирусной популяции от сероконверсии (первый круг). Вертикальное смещение кругов показывает степень дивергенции вирусной популяции (divergence) от предкового штамма (founder strain). Затенения соответствуют пропорции вирусной популяции, представленной X4-генотипом. Вертикальные линии (начиная с левой стороны схемы) соответствуют: окончанию стадии острой инфекции; пику вирусного разнообразия; стабилизации дивергенции от предкового штамма; развитию СПИДа. В начале поздней фазы дивергенции (роста разнообразия) количество X4-вариантов ВИЧ начинает снижаться. Эта фаза проявляется нарушением гомеостаза Т-клеток. Количество CD4+ T-клеток снижается до уровня <200 клеток/мм3, появляются симптомы выраженного поражения клеточной системы иммунитета, болезнь переходит в стадию СПИДа. Теперь разнообразие вариантов вируса идет на убыль, так как иммунная система истощена и уже не способна раскручивать маховик его эволюции. По [109]
Рисунок 10. Схематическое изображение развития ВИЧ-инфекции у умеренных прогрессоров. Диаметры кругов приблизительно соответствуют разнообразию (diversity) вирусной популяции от сероконверсии (первый круг). Вертикальное смещение кругов показывает степень дивергенции вирусной популяции (divergence) от предкового штамма (founder strain). Затенения соответствуют пропорции вирусной популяции, представленной X4-генотипом. Вертикальные линии (начиная с левой стороны схемы) соответствуют: окончанию стадии острой инфекции; пику вирусного разнообразия; стабилизации дивергенции от предкового штамма; развитию СПИДа. В начале поздней фазы дивергенции (роста разнообразия) количество X4-вариантов ВИЧ начинает снижаться. Эта фаза проявляется нарушением гомеостаза Т-клеток. Количество CD4+ T-клеток снижается до уровня <200 клеток/мм3, появляются симптомы выраженного поражения клеточной системы иммунитета, болезнь переходит в стадию СПИДа. Теперь разнообразие вариантов вируса идет на убыль, так как иммунная система истощена и уже не способна раскручивать маховик его эволюции. По [109]
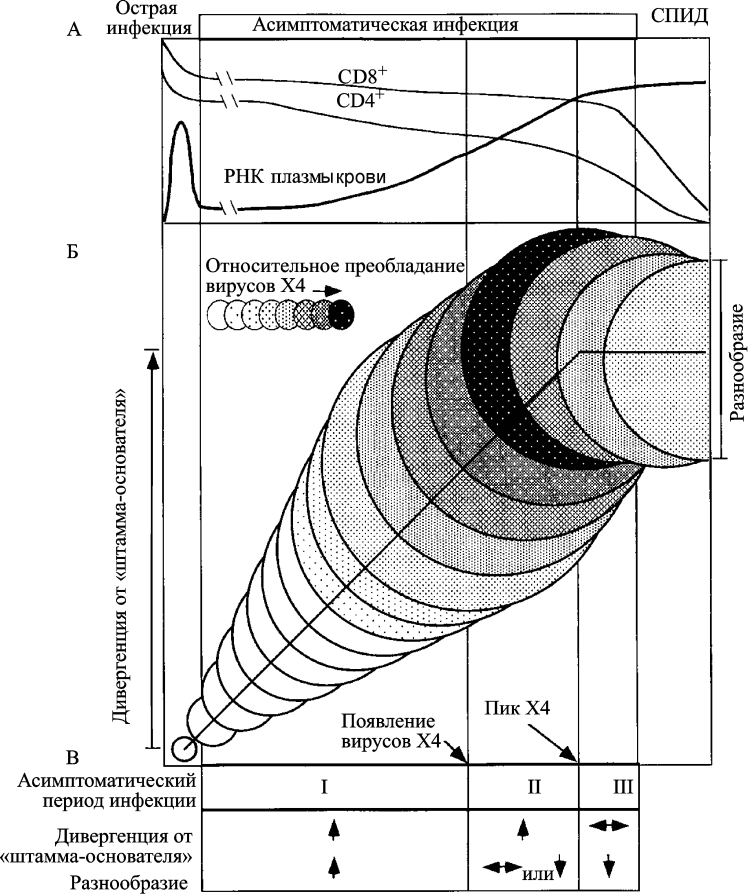
Приведенные Shankarappa et al. [109] данные показывают, что в раннюю фазу инфекции развиваются оба процесса; промежуточная фаза характеризуется непрерывным увеличением дивергенции ВИЧ, но стабилизацией или даже снижением его разнообразия; поздняя фаза проявляется снижением темпа или даже стабилизацией процессов дивергенции и формирования разнообразия вируса. Результатом работы такого механизма являются:
1)массивное распространение ВИЧ по фагоцитирующим клеткам;
2)повышение его вирулентности за счет отбора вариантов, тропных к рецептору CXCR4.
По данным Zhang et al. [110], увеличение генетического разнообразия вируса субтипа С у детей зависит от антител с широким нейтрализующим действием. Чем выше титр таких антител, тем больше на данный момент времени вирусы различаются между собой. То, что ВИЧ меняется не сам, а его в ходе инфекционного процесса меняет иммунная система с помощью феномена ADE и специфических антител, выглядит странно только в контексте медицинского подхода к пониманию ВИЧ/СПИД-пандемии. Но это проблема гораздо шире.
ВИЧ относится к семейству Retroviridae. Вирусы этого семейства интегрируют свою ДНК-копию (провирус) с геномом хозяина в единую молекулу ДНК. Если ретровирус становится частью генома вида, то вид считается прошедшим через эндогенизацию. Эндогенные ретровирусы активны в геноме вида и его видов–потомков до 6 млн лет. Они передаются вертикально, инициируя наращивание его генетического материала образованием своих новых копий, усложняя геном образованием новых экзонов из интронов и/или увеличивая количество генов, подвергающихся альтернативному сплайсингу [111–116]. По своей сути этот процесс представляет один из механизмов эволюции видов. Эволюционное прошлое иммунной системы многоклеточных организмов свидетельствует о закреплении за ней естественным отбором резервуарной роли по отношению к ретровирусам. Благодаря клеткам иммунной системы происходят размножение и накопление в популяции вида экзогенных ретровирусов до какой-то критической массы, которая позволяет некоторым из них эндогенизироваться в зародышевой линии отдельных особей инфицированного вида и в дальнейшем передаваться вертикально, меняя его эволюционную траекторию в течение миллионов лет. Этот процесс приобрел глобальный характер среди нашего вида под маской ВИЧ/СПИД-пандемии [111].
Феномен ADE, развивающийся на фоне сенсибилизации, вызваннойвакцинацией. Осложнения после вакцинации, возникающие как следствие феномена ADE, до настоящего времени не стали объектом системных исследований, поэтому сведения о них носят разрозненный характер (табл. 4).
Таблица 4. Феномен ADE, развивающийся как ответ на вакцинацию*
|
Вирус (семейство) |
Доказан в условиях in vitro |
Доказан в условиях in vivo |
|
РНК-вирусы |
|
|
|
Вирус гриппа А (Orthomyxoviridae) |
+ |
+ |
|
Респираторный синтициальный вирус (Paramyxoviridae) |
+ |
+ |
|
Вирус кори (Paramyxoviridae) |
+ |
+ |
|
Вирус бешенства (Rhabdoviridae) |
+ |
+ |
|
Вирус кошачьего инфекционного перитонита (Coronaviridae) |
+ |
+ |
|
Вирус свиного репродуктивного и респираторного синдрома (Coronaviridae) |
+ |
+ |
|
Вирус обезьяньей геморрагической лихорадки (Coronaviridae) |
+ |
+ |
|
ВИЧ (Retroviridae) |
+ |
? |
|
Вирус лошадиной инфекционной анемии (Retroviridae) |
+ |
+ |
|
Вирус артрита коз (Retroviridae) |
+ |
+ |
|
ДНК-вирусы |
|
|
|
Вирус алеутской болезни норок (Parvoviridae) |
+ |
+ |
*По [55].
Феномен ADE у ранее вакцинированного человека может быть связан:
1)с неполноценной иммунизацией;
2)особенностями взаимодействия возбудителя инфекционной болезни с иммунной системой человека;
3)особенностями эпидемического очага, в котором проводится вакцинация.
Неполноценная иммунизация. Причинно-следственная связь ADE с неполноценной иммунизацией подробно изучена на примерах инактивированной коревой вакцины и инактивированной вакцины против респираторного синцитиального вируса (respiratory syncytial virus, RSV) [117, 118]. Обе вакцины получают путем инактивации вирусов формальдегидом. C начала 1960-х гг., т.е. после начала массовых иммунизаций населения против кори вакцинами, инактивированными формалином[22], среди вакцинированных людей отмечаются случаи так называемой атипичной кори (кори, протекающей в тяжелой форме). Iankov et al. [81] показали, что в основе ее развития лежит феномен FcR–ADE, вызываемый антителами к гемагглютинину вируса (поверхностный белок Н).
Установлено, что антитела, полученные в отношении антигенных белков вирусов кори и RSV, инактивированных формальдегидом, обладают сниженной протективной способностью по сравнению с антителами, полученными в отношении этих же антигенов живых вакцин. Это вызвано тем, что подвергнутые обработке формалином антигенные белки имеют увеличенное количество активных карбонильных групп, что ведет к нарушению третичной структуры эпитопов [119, 120].
ADE как феномен, характерный для взаимодействия возбудителя инфекционной болезни с иммунной системой человека. Если ADE развивается в ходе инфекционного процесса, то есть основание считать, что феномен будет иметь место у вакцинированных людей и животных, если они будут заражены вирусом, против которого их вакцинировали (табл. 3, 4).
Показательны результаты экспериментов с вакцинами, разрабатываемыми для специфической профилактики ретровирусных инфекций у животных — инфекционной анемии лошадей и иммунодефицита кошек. Также они имели цель моделирования стратегий вакцинации против ВИЧ. Хотя эти эксперименты выполнены еще в 1990-х гг., они до сих пор не вызвали интереса у разработчиков ВИЧ-вакцин.
Инфекционная анемия лошадей вызывается вирусом инфекционной анемии лошадей (Equine infectious anemia virus, EIAV). Болезнь носит нециклический характер, проявляется синдромами лихорадки, анорексии, анемии, выздоровления не наступает. Показано серьезное обострение болезни при заражении EIAV вакцинированных лошадей и пони, если в их сыворотке присутствовали антитела, индуцированные введением вакцины. Issel at al. [71] использовали виремию как критерий тяжести болезни и продемонстрировали, что вакцинация инактивированной цельновирионной вакциной не может предотвратить развитие виремии и клинических симптомов болезни у животного, которому введен вирулентный штамм вируса. В экспериментах по заражению гетерологичным штаммом вируса животных, вакцинированных высокоочищенным оболочечным гликопротеином вируса, также не удавалось предотвратить ни виремию, ни развитие клинических симптомов болезни. В последующем Wang et al. [72] провели масштабные эксперименты на пони и лошадях по оценке защитной эффективности рекомбинантной вакцины, полученной на основе поверхностного гликопротеина EIAV. Результаты экспериментов показали усиление инфекции у всех предварительно вакцинированных животных.
Ретровирус, возбудитель иммуно–дефицита кошек (Feline immunodeficiency virus, FIV), после инфицирования кошек, вакцинированных оболочечным рекомбинантным белком этого вируса, обнаруживался в их крови даже раньше, чем у невакцинированных животных [75]. В сходных исследованиях, выполненных с различными рекомбинантными FIV–вакцинами, было установлено, что в крови животных в ответ на вакцинацию обнаруживаются антитела к оболочечному белку (env) FIV, плохо нейтрализующие вирус в условиях in vitro. У вакцинированных животных вирусная нагрузка была значительно большей, чем у невакцинированных. При росте титров антител к коровому белку (core protein) FIV у кошек имело место усиление клинических признаков болезни [73, 121, 122].
Сходные результаты получены в экспериментах на людях по изучению протективного эффекта ВИЧ-вакцины, проведенных в Южной Африке фирмой Merck. Из 741 вакцинированного добровольца 24 впоследствии заразились ВИЧ. В другой группе добровольцев, получивших плацебо, 21 из 762 участников также были инфицированы. Эксперимент, по результатам больше похожий на преступление, был досрочно прекращен [123]. Исследователи из Merck предпочли не интересоваться тем, сколько ВИЧ-инфицированных будет в обеих группах через три, пять и более лет. Их не заинтересовало сравнение данных о том, как быстро болезнь переходит в стадию СПИДа у вакцинированных и невакцинированных. Как будто, прекратив «эксперимент», можно прекратить развитие ADE в случае контакта иммунной системы вакцинированного ВИЧ-вакциной африканца с ВИЧ[23].
Косвенные доказательства развития феномена ADE в ходе туберкулезного процесса [63] хорошо согласуются с наблюдениями Норейко [126], показавшего, что у людей, вакцинированных вакциной БЦЖ (Bacillus Calmette–Gurin, BCG), вторичные формы туберкулеза склонны к прогрессированию с развитием таких осложнений, как деструкция легочной ткани с бактериовыделением и бронхогенной диссеминацией. Однако с этой точки зрения диссеминация туберкулезного процесса им не рассматривалась, так как феномен ADE не известен клиницистам.
Особенности эпидемического очага, в котором проводится вакцинация. Wallace et al. [83] в опытах на мышах установили, что антитела к вирусу японского энцефалита в субнейтрализующих концентрациях увеличивают вирусемию и смертность среди мышей, зараженных вирусом энцефалита долины Мюррей (MVEV). На основании этих данных они предположили, что феномен ADE может способствовать замене одного эпидемического процесса другим. Исследователи считают, что программы по вакцинации населения против вируса японского энцефалита в тех районах, где одновременно с ним циркулирует и MVEV, могут способствовать развитию эпидемии энцефалита долины Мюррей.
Роль ADE в эпидемических, инфекционных и поствакцинальныхпроцессах. В общем виде ее можно представить следующим образом:
1)если ADE развивается на фоне сенсибилизации, вызванной предшествующим инфекционным процессом, то в эпидемических процессах он проявится усилением тяжести инфекционного процесса у отдельных пациентов, ранее переболевших или вакцинированных, и большим количеством осложнений и летальных исходов при повторении эпидемической вспышки;
2)если ADE развивается без предварительной сенсибилизации иммунной системы, то он будет играть основную роль в патогенезе инфекционной болезни;
3)при развитии ADE в ходе персистирующего инфекционного процесса его роль будет заключаться в усилении тяжести инфекционного процесса, селекции наиболее опасных штаммов возбудителя инфекционной болезни с последующим вовлечением их в новые эпидемические цепочки;
4)ADE у людей, вакцинированных неполноценными вакцинами, может проявиться тяжелым течением болезни при инфицировании возбудителем, в отношении которого проводилась вакцинация. Если ADE развивается в ходе инфекционного процесса, вызванного возбудителем инфекционной болезни, в отношении которого проводилась вакцинация, либо пациенту вводился специфический иммуноглобулин, ADE может развиться после его заражения этим возбудителем в эпидемическом очаге. В случае одновременной циркуляции в популяции людей нескольких возбудителей инфекционных болезней, когда антитела к одному из вирусов способны в субнейтрализующих концентрациях увеличивать размножение другого, при наличии механизма передачи возбудителя болезни между людьми ADE может способствовать замене одного эпидемического процесса другим.
***
Феномены антигенного импринтинга и антитело-зависимого усиления инфекции известны ученым уже более 50 лет. Они определяют тяжесть эпидемического и инфекционного процессов и эффективность проведенной вакцинации. В клинической практике осложнения, связанные с этими феноменами, встречаются чаще, чем принято думать, но их не интерпретируют правильно даже опытные клиницисты из-за пробелов в базовых представлениях об иммунитете. Игнорирование антигенного импринтинга и антитело-зависимого усиления инфекции при разработке новых вакцин, их доклинических и клинических исследованиях и проведении массовых вакцинаций населения представляет собой профанацию эпидемиологии и иммунологии, выгодную лишь недобросовестным производителям устаревших вакцин. Профессорско-преподавательский состав медицинских вузов должен понимать и то, что подготовка специалистов, имеющих неполное представление о работе иммунной системы, особенно в такой сфере, как борьба с инфекционными болезнями, создает условия для различных злоупотреблений, тормозит развитие отечественной эпидемиологии и иммунологии, и отдает приоритет новых научных открытий зарубежным ученым.
Литература
1. Богомолов БП. Инфекционные болезни: неотложная диагностика, лечение и профилактика. М.: 2007.
2. Борисов Л.Б. Медицинская микробиология, вирусология, иммунология. М.: 2001.
3. Воробьев А.А. Не подводя черты. М.: 2003.
4. Воробьев А.А, Быков АС, Пашков ЕП, Караулов АВ. Микробиология. М.: 1992.
5. Галактионов В.Г. Эволюционная иммунология. М.: 2005.
6. Черкасский Б.Л. Руководство по общей эпидемиологии. М.: 2001.
7. Медуницын НВ, Покровский ВИ. Основы иммунопрофилактики и иммунотерапии инфекционных болезней. М.: 2005.
8. Супотницкий МВ. Феномен антигенного импринтинга при доклиническом изучении иммунобиологических лекарственных препаратов. В кн.: Миронов АН ред. Руководство по проведению доклинических исследований лекарственных средств (иммунобиологические лекарственные препараты). Часть вторая. М.: Гриф и К; 2012. С. 185–90.
9. Миронов АН, Супотницкий МВ, Лебединская ЕВ. Феномен антитело-зависимого усиления инфекции у вакцинированных и переболевших. Биопрепараты 2013; (3): 12–25.
10. Супотницкий МВ. Феномен антителозависимого усиления инфекции при доклиническом изучении иммунобиологических лекарственных препаратов. В кн.: Миронов АН ред. Руководство по проведению доклинических исследований лекарственных средств (иммунобиологические лекарственные препараты). Часть вторая. М.: Гриф и К; 2012. C. 177–85.
11. Мац АН. Первородный антигенный грех (original antigenic sin) как антипрививочная теологическая аллюзия. [Интернет]. 2009 [cited 2014 Jan 21]. Available from: http://copy.yandex.net/?lang=ru&fmode=envelope&tld=ru&text=%D0%BC%D0%25B0%D1%86%20%D0%BF%D0%B5%D1%80%D0%B2%D0%B8%D1%87%D0%BD%D1%8B%D0%B9%20%D0%B0%D0%BD%D1%82%D0%B8%D0%B3%D0%B5%D0%BD%D0%BD%D1%8B%D0%B9%20%D0%B3%D1%80%D0%B5%D1%85%20%D1%81%D1%83%D0%BF%D0%BE%D1%82%D0%BD%D0%B8%D1%86%D0%BA%D0%B8%D0%B9&url=http%3A%2F%2Fwww.vnpoemp.ru%2Ffile%2Fdoc%2Fdoc_091203.doc&lr=213&l10n=ru&mime=doc&sign=d768252182e77aaaf30516353b580f5f&keyno=0.
12. Райт АЕ. Основы вакцинотерапии (теория опсонинов). Спб.: 1908.
13. Francis T. Influenza. The newe acquayantance. Ann Int Med. 1953; 399(3): 201–21.
14. Горбунова АС. Грипп. В кн.: Руководство по микробиологии, клинике и эпидемиологии инфекционных болезней. М.: Медицина; 1966. Т. VIII; 13–60.
15. Ma J, Dushoff J, Earn DJ. Age-specific mortality risk from pandemic influenza. J Theor Biol. 2011; 288: 29–34. doi: 10.1016/j.jtbi.2011.08.003.
16. Kim JH, Skountzou I, Compans R, Jacob J. Original antigenic sin responses to influenza viruses. J Immunol. 2009; 183: 3294–301.
17. Павлович СА. Основы иммунологии. М.: 1997.
18. Davenport FM, Hennessy AV, Francis T. Epidemiologic and immunologic significance of age distribution of antibody to antigenic variants of influenza virus. J Exp Med. 1953; 98: 641–56.
19. Francis T. The current status of the control of influenza. Ann. Int. Med. 1955; 43: 534–8.
20. Davenport FM, Hennessy AV. A serologic recapitulation of past experience with influenza A; antibody response to monovalent vaccine. J Exp Med. 1956; 104: 85–97.
21. Литвинова ОМ, Лузянина ТЯ. Этиология гриппа. В кн. Грипп. Руководство для врачей. СПб.: Гиппократ; 2001. С. 7–30.
22. Marine WM, Thomas JE. Antigenic memory to influenza A viruses in man determined by monovalent vaccines. Postgraduate Medical Journal 1979; 55: 98–108.
23. Masurel N, Drescher J. Production of highly cross-reactive hemagglutination-Inhibiting influenza antibodies in ferrets. Infection and Immunity 1976; 13(4): 1023–9.
24. Карпухин ГИ, Швецова ЕГ, Малышева АМ. Итоги многолетнего опыта изучения эффективности экстренной профилактики гриппа ремантадином в эпидемиологических наблюдениях. В кн.: Проблемы гриппа и острых респираторных болезней. Л.: 1979. С. 24–8.
25. Angelova L, Shvartsman Y. Original antigenic sin to influenza in rats. Immunology 1982; 46. 183–8.
26. Couch RB, Webster RG, Kasel TR, Cate TR. Efficacy of purified influenza subunit vaccines and relation to the major antigenic determinants on the hemagglutinin molecule. J Infect Dis. 1979; 140: 553–9.
27. Keitel WA, Cate TR, Couch RB, Huggins LL, Hess KR. Efficacy of repeated annual immunization with inactivated influenza virus vaccines over a five year period. Vaccine 1997; 15, 1114–22.
28. Smith DJ, Forrest S, Ackley DH, Perelson AS. Variable efficacy of repeated annual influenza vaccination. Proc Natl Acad Sci U S A 1999; 96(24): 14001–6
29. Klenerman P, Zinkernagel RM. Original antigenic sin impairs cytotoxic T lymphocyte responses to viruses bearing variant epitopes. Nature 1998; 394: 482–5. DOI: 10.1038/28860.
30. Duangchindaa T, Dejnirattisaia W, Vasanawathanac S, Limpitikuld W, Tangthawornchaikulb N, Malasitb P. et al. Immunodominant T-cell responses to dengue virus NS3 are associated with DHF. Proc Natl Acad Sci U S A 2010; 107(39): 16922–7.
31. Crill WD, Hughes HR, Trainor NB, Davis BS, Whitney MT, Chang GJ. Sculpting humoral immunity through dengue vaccination to enhance protective immunity. Frontiers in Immunology 2012; 3: Art. 334. doi: 10.3389/fimmu.2012.00334.
32. Midgley CM, Bajwa-Joseph M, Vasanawathana S, Limpitikul W, Wills B, Flanagan A et al. An in-depth analysis of original antigenic sin in Dengue virus infection. J Virol. 2011; 85 (1): 410–21.
33. Kim JH, Davis WG, Sambhara S, Jacob J. Strategies to alleviate original antigenic sin responses to influenza viruses. Proc Natl Acad Sci U S A 2012; 109(34): 13751–6.
34. Choi YA, Baek YH, Kang W, Nam SJ, Lee J, You S et al. Reduced antibody responses to the pandemic (H1N1) 2009 vaccine after recent seasonal influenza vaccination. Clin Vaccine Immunol. 2011; 18(9): 1519–23.
35. Zhang X, He J, Li L, Zhu X, Ke C, Ni H et al. Serologic survey of the pandemic H1N1 2009 virus in Guangdong province, China: a cross sectional study. PLoS ONE 2011; 6(8): e23034. doi:10.1371/journal.pone.0023034
36. Adalja AA, Henderson DA. Original antigenic sin and pandemic (H1N1) 2009. Emerging Infectious Diseases 2010; 16(6): 1028–9.
37. Chowell G, Bertozzi SM, Colchero АM, Lopez-Gatell H, Alpuche-Aranda C, Hernandez M, Miller MA. Severe respiratory disease concurrent with the circulation of H1N1 influenza. N Engl J Med 2009; 361: 674-9.
38. Reichert T, Chowell G, Nishiura H, Christensen RA, McCullers JA. Does glycosylation as a modifier of original antigenic sin explain the case age distribution and unusual toxicity in pandemic novel H1N1 influenza? BMC Infectious Diseases 2010; 10(5). Available from: http://www.biomedcentral.com/1471-2334/10/5.
39. Skowronski DM, DeSerres G, Crowcroft N et al. Association between the 2008–09 seasonal influenza vaccine and pandemic H1N1 illness during spring–summer 2009: four observational studies from Canada. PLoS Med. 2010; 7(Is.4): e1000258. doi:10.1371/journal.pmed.1000258.
40. Janjua NZ, Skowronski DM, Hottes TS, Osei W, Adams E, Petric M et al. Seasonal influenza vaccine and increased risk of pandemic A/H1N1‐related illness: first detection of the association in British Columbia, Canada. Clin Infect Dis. 2010; 51(9): 1017–27.
41. Monsalvo AC, Batalle1 JP, Lopez MF, Krause JC, Klemenc J, Zea J et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat Med. 2011; 17(2): 195–9. doi:10.1038/nm.2262.
42. Parsons MS, Muller S, Kohler H, Grant MD, Bernard NF. On the benefits of sin Can greater understanding of the 1F7-idiotypic repertoire freeze enhance HIV vaccine development? Human Vaccines & Immunotherapeutics 2013; 9(7): 1532–8.
43. Nara PL, Garrity RR, Goudsmit J. Neutralization of HIV-1: a paradox of humoral proportions. FASEB J. 1991; 5: 2437–55.
44. Narayan O, Griffin DE, Clements JE. Virus mutation during «slow infection»: temporal development and characterization of mutants of visna virus recovered from sheep. J Gen Virol. 1978; 41: 343–52.
45. Kono Y, Kobayashi K, Fukunaga Y. Serological comparison among various strains of equine infectious anemia virus. Arch Gesamte Virusforsch. 1971; 34: 202–8.
46. Locher CP, Grant RM, Collisson EA, Reyes-Terbn G, Elbeik T, Kahn JO et al. Antibody and cellular immune responses in breakthrough infection subjects after HIV type 1 glycoprotein 120 vaccination. AIDS Res Hum Retroviruses 1999; 15: 1685–9. Available from: http://dx.doi.org/10.1089/088922299309720.
47. Muller S, Wang H, Silverman GJ, Bramlet G, Haigwood N, Khler H. B-cell abnormalities in AIDS: stable and clonally-restricted antibody response in HIV-1 infection. Scand J Immunol. 1993; 38: 327–34; PMID:7692591. Available from: http://dx.doi.org/10.1089/088922299309720
48. Pleass RJ, Ogun SA, McGuinness DH, van de Winkel JG, Holder AA, Woof JM Novel antimalarial antibodies highlight the importance of the antibody Fc region in mediating protection. Blood 2003; 102(13): 4424-30.
49. Wipasa J, Xu H, Liu X, Hirunpetcharat C, Stowers A, Good MF, Effect of Plasmodium yoelii exposure on vaccination with the 19-kilodalton carboxyl terminus of merozoite surface protein 1 and vice versa and implications for the application of a human malaria vaccine. Infection and Immunity 2009; 77(2): 817–24.
50. Flipse J, Wilschut J, Smit JM. Molecular mechanisms involved in antibody-dependent enhancement of Dengue virus infection in humans. Traffic 2013; 14: 25–35.
51. Halstead SB, Rojanasuphot S, Sangkawibha N. Original antigenic sin in dengue. Am J Trop Med Hyg. 1983; 32: 154–6.
52. Brown SA, Surman SL, Sealy R, Jones BG, Slobod KS, Branum K. et al. Heterologous prime-boost HIV-1 vaccination regimens in pre-clinical and clinical trials. Viruses 2010; 2(2): 435–67. doi:10.3390/v2020435.
53. Hawkes RA. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls. Aust J Exp Biol Med Sci. 1964; 43: 465–82.
54. Hawkes RA, Lafferty KJ. The enhancement of virus infectivity by antibody. Virology 1967; 33: 250–61.
55. Halstead SB, Mahalingam PS, Marovich MA, Ubol S, Mosser DM. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis. 2010; 10(10): 712–22.
56. Halstead SB, Chow J, Marchette NJ. Immunologic enhancement of Dengue virus replication. Nat New Biol. 1973; 243: 24–6.
57. Thomas HI, Wilson S, O'Tolle CM, Lister CM, Saeed AM, Watkins RP, Morgan-Capner P. Differential maturation of avidity of IgG antibodies to gp41, p24 and p17 following infection with HIV-1. Clin Exp Immunol. 1996; 103: 185–91.
58. Henchal EA, McCown JM, Burke DS, Seguin MC, Brandt WE. Epitopic analysis of antigenic determinants on the surface of Dengue-2 virions using monoclonal antibodies. Am J Trop Med Hyg. 1985; 34: 164–9.
59. Kreil TR, Eibl MM. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model. J Virol. 1997; 71(4): 2921–7.
60. Tirado SM., Yoon KJ. Antibody-dependent enhancement of virus infection and disease. Viral Immunol. 2003; 16: 69–86.
61. Yoong P. Enhancement of bacterial virulence by antibody neutralization of immune-activating toxins. Virulence 2010; 1(5): 409–13.
62. He X, Sun X, Wang J, Wang X, Zhang Q, Tzipori S, Feng H. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Inf Immunol. 2009; 77(6): 2294–303.
63. Maglione PJ, Xu J, Casadevall A, Chan J. Fc gamma receptors regulate immune activation and susceptibility during Mycobacterium tuberculosis infection. J Immunol. 2008; 180: 3329–38.
64. Reimer LG. Q Fever. Clin. Microbiol. Rev. 1993; 6(3): 193–8.
65. Shannon J, Heinzen R. Adaptive immunity to the obligate Intracellular pathogen Coxiella burnetii. Immunol Res. 2009; 43(1–3): 138–48.
66. Константинова НА. Иммунные комплексы и повреждение тканей. М.: 1996.
67. Заболотный ДК. Пустулезная форма чумы. Русский архив патологии 1899; VIII: 239–42.
68. Takada A, Feldmann H, Ksiazek TG, Kawaoka Y. Antibody-dependent enhancement of Ebola virus infection. J Virol. 2003; 77(13): 7539–44.
69. Homsy J, Meyer M, Tateno M, Clarkson S, Levy JA. The Fc and not CD4 receptor mediates antibody enhancement of HIV infection in human cells. Science 1989; 16(244): 1357–60.
70. Robinson WE, Montefiori DC, Mitchell WM. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection. Lancet 1988; 9(1): 790–4.
71. Issel CJ, Horohov DW, Lea DF, Adams WV, Hagius SD, McManus JM. et al. Efficacy of inactivated whole-virus and subunit vaccines in preventing infection and disease caused by equine infectious anemia virus. J Virol. 1992; (66): 3398–408.
72. Wang SZ, Rushlow KE, Issel CJ, Cook RF, CooSJ, Raabe ML et al. Enhancement of EIAV replication and disease by immunization with a baculovirus-expressed recombinant envelope surface glycoprotein. Virol. 1994; 199: 247–51.
73. Hosie MJ, Osborne R, Reid G, Neil JC, Jarrett O. Enhancement after feline immunodeficiency virus vaccination. Vet Immunol Immunopathol. 1992; 35: 191–7.
74. Takano T, Kawakami S, Yamada S, Satoh R, Hohdatsu T. Antibody-dependent enhancement occurs upon re-infection with the identical serotype virus in feline infectious peritonitis virus infection. J Vet Med Sci. 2008; 70(12): 1315–21.
75. Radkowski MT, Laskus T, Goch A, Slusarczyk J. Affinity of anti-GP41 antibody in patients infected with human immunodeficiency virus type I. Eur J Clin Invest. 1993; 23(8): 455–8.
76. Takada A, Watanabe S, Okazak K, Kida H, Kawaoka Y. Infectivity enhancing antibodies to Ebola virus glycoprotein. J Virol. 2001 75(5): 2324–30.
77. Nakayama E, Tomabechi D, Matsuno K, Kishida N, Yoshida N, Feldmann H, Takada A. Antibody-dependent enhancement of Marburg virus infection. J Infect Dis. 2011; 204: 978–85.
78. Meyer K, Ait-Goughoulte M, Keck Zhen-Yong, Foung S, Ray R. Antibody-dependent enhancement of hepatitis C virus infection. J Virol. 2008; 829(5): 2140–9.
79. Barrett ADT, Gould A. Antibody-mediated early death in vivo after infection with Yellow fever virus. J Gen Virol. 1986 (67): 2539–42.
80. Goulld EA, Buckley A. Antibody-dependent enhancement of Yellow Fever and Japanese encephalitis virus neurovirulence. J Gen Virol. 1989; 70: 1605–8.
81. Iankov ID, Pandey M, Harvey M, Griesmann GE, Federspiel MJ, Russell SJ. Immunoglobulin G antibody-mediated enhancement of measles virus infection can bypass the protective antiviral immune response. J Virol 2006; 80(17): 8530–40.
82. Mehlhop E, Ansarah-Sobrinho C, Johnson S, Engle M, Fremont DH, Pierson TC, Diamond MS. C1q Inhibits antibody-dependent enhancement of Flavivirus infection in vitro and in vivo in an IgG subclass specific manner. Cell Host Microbe. 2007; 2(6): 417–26.
83. Wallace MJ, Smith DW, Broom AK, Mackenzie JS, Hall RA, Shellam GR, McMinn PC. Antibody-dependent enhancement of Murray Valley encephalitis virus virulence in mice. J Gen Virol. 2003; 84(7): 1723–8.
84. Balsitis SJ., Williams KL., Lachica R, Flores D, Kyle JL, Mehlhop E, Johnson S et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 2010; 12: doi: 10.1371/journal.ppat.1000790.
85. Han J-F, Cao R, Deng Y, Tian X, Jiang T, Qin ED, Qin CF. Antibody dependent enhancement infection of Enterovirus 71 in vitro and in vivo. Virol J; 2011: 8. Available from http://www.virologyj.com/content/8/1/106.
86. Blancou J, Andrai B, Andrai L. A model in mice for the study of the early death phenomenon after vaccination and challenge with rabies virus. J Gen Virol. 1980; 50: 433–5.
87. King AA, Sands JJ, Porterfield JS. Antibody-mediated enhancement of rabies virus infection in a mouse macrophage cell line (P388D1). J Gen Virol. 1984; 65: 1091–3.
88. Prabhakar BS, Nathanson N. Acute rabies deaths mediated by antibody. Nature 1981; 290: 590–1.
89. Porter AD, Larsen AE, Porter HG. The pathogenesis of Aleutian disease of mink. II. Enhancement of tissue lesions following the administration of a killed virus vaccine or passive antibody. J Immunol. 1972; 109: 1–7.
90. Lidbury BA, Mahalingam S. Specific ablation of antiviral gene expression in macrophages by antibody-dependent enhancement of Ross River virus infection. J Virol. 2000; 74: 8376–81.
91. Linn ML, Aaskov G, Suhrbier A. Antibody-dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis. J Gen Virol. 1996; 77: 407–11.
92. Супотницкий МВ. Пандемия «испанки» 1918–1920 гг. в контексте других гриппозных пандемий и «птичьего гриппа». Медицинская картотека 2006; (11): 31–34; (12): 15–25, 28–30; 2007; (1): 16–22. Available from https://www.supotnitskiy.ru/stat/stat51.htm.
93. Tsai YT, Chang SY, Lee CN, Kao CL. Human TLR3 recognizes Dengue virus and modulates viral replication in vitro. Cell Microbiol. 2009; 11: 604–15.
94. Ubol S, Chareonsirisuthigul T, Kasisith J, Klungthong C. Clinical isolates of Dengue virus with distinctive susceptibility to nitric oxide radical induce differential gene responses in THP-1 cells. Virology 2008; 376: 290–6.
95. Dejnirattisai W, Jumnainsong A, Onsirisakul N. Cross-reacting antibodies enhance dengue virus infection in humans. Science 2010; 328: 745–8.
96. Mathew A, West K, Kalayanarooj S, Gibbons RV, Srikiatkhachorn A, Green S et al. B-cell responses during primary and secondary dengue virus infections in humans. J Infect Dis. 2011; 204: 1514–22.
97. Kurosu T. Quasispecies of dengue virus. Trop. Med. and Health. 2011; 39(4): 29–36.
98. Купер Э. Сравнительная иммунология. М.: 1980.
99. Zellweger RM, Prestwood TR, Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe. 2010; 7: 128–39.
100. Chaturvedi UC, Raghupathy R, Pasca AS. Shift from a Th1-type response to Th1-type in dengue haemorrhagic fever. Curr Sci. 1999; 76: 63–9.
101. Ubol S, Halstead SB. How innate immune mechanisms contribute to antibody-enhanced viral infections. Clin Vac Immunol. 2010; 17(12): 1829–35.
102. Takada A, Ebihara H, Feldmann H, Geisbert TW, Kawaoka Y. Epitopes required for antibody-dependent enhancement of Ebola virus infection. J Infec Dis. 2007; 196: 347–56.
103. Vennema H, De Groot R, Harbour D, Dalderup M, Gruffydd-Jones T, Horzinek MC, Spaan WJ. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J Virol. 1990; 64: 1407–9.
104. Vennema H, Poland A, Foley J, Pedersen NC. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology. 1998; 243(1): 150–7.
105. Fust G. Enhancing antibodies in HIV infection. Parasitology 1997; 115: Suppl: S. 127–40.
106. Takeda A, Tuazon CU, Ennis FA. Antibody-enhanced infection by HIV-1 via Fc receptor-mediated entry. Science 1988; 242(4878): 580–3.
107. Burton DR, Stanfield RL, Wilson IA. Antibody vs. HIV in a clash of evolutionary titans. Proc Natl Acad Sci USA. 2005;102(42): 14943–1494.
108. Frost S, Wrin T, Smith DM. Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection. Proc Natl Acad Sci USA. 2005; 102(51): 18514–9.
109. Shankarappa R, Margolick JB, Gange SJ. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol. 1999; 73(12): 10489–502.
110. Zhang H, Hoffmann F, He J, Xiang He, Chipepo Kankasa, Ruth Ruprech et al. Evolution of subtype C HIV-1 Env in a slowly progressing Zambian infant. Retrovirology. 2005. Available from: http://www.retrovirology.com/content/2/1/6.
111. Супотницкий МВ. Эволюционная патология. М.: 2009.
112. Bannert N, Kurth R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA. 2004; 101(Suppl. 2):14572–9.
113. Coffin JM. Evolution of retroviruses: fossils in our DNA. Proceed. Amer. Philosoph. Soc. 2004; 148(3): 264–80.
114. Costas J, Naverira H. Evolutionary history of the human endogenous retrovirus family ERV9. Mol Biol Evol. 2000; 17(2): 320–330.
115. Богадельников ИВ, Смирнов ГИ. Современные представления о течении инфекционных и эпидемических процессов. Новости медицины и фармации. 2013; 16: 20–1.
116. Богадельников ИВ, Смирнов ГИ. Особенности течения инфекционных и эпидемических процессов в настоящее время. Актуальная инфектология. 2013; (1): 68–72.
117. Fulginiti FA, Eller JJ, Downie AW, Kempe CH. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines. JAMA. 1967 (202): 1075–80.
118. Kim HW, Canchola JG, Brandt CD. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine. Am. J. Epidemiol. 1969; 89: 422–34.
119. Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. 2009; 15: 34–41.
120. Moghaddam A, Olszewska W, Wang B, Tregoning JS, Helson R, Sattentau QJ, Openshaw PJ. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines. Nat Med. 2006; 12905–7.
121. Baldinotti FD, Matteucci P, Mazzetti P, Giannelli C, Bandecchi P, Tozzini F, Bendinelli M. Serum neutralization of feline immunodeficiency virus is markedly dependent on passage history of the virus and host system. J Virol. 1994; 74: 10834–7.
122. Huisman W, Karlas KH, Siebelink KH, Huisman RC, de Ronde A, Francis MJ. et al. Feline immunodeficiency virus subunit vaccines that induce virus neutralizing antibodies but no protection against challenge infection. Vaccine 1998;16: 181–7.
123. Vaccination and Enrollment Are Discontinued in Phase II Trials of Merck's Investigational HIV Vaccine Candidate. News Release. Available from http://www.hvtn.org/pdf/FINAL_HIV_Vaccine_Press_Release.pdf.
124. Мiронов ОМ, Супотнiцький МВ, Лебединська ОВ. Феномен антитiлозалежного посилення iнфекцii у вакцинованих i перехворiлих. Iнфекцiйнi хвороби 2013; (4): 86–91. 2014; (1): 69–79.
125. Broker M, Kollaritsch H. After a tick bite in a tick-borne encephalitis virus endemic area: current positions about post-exposure treatment. Vaccine 2008; 26(7): 863-8.
126. Норейко БВ. Иммунологические аспекты фтизиатрии. Новости медицины и фармации (Донецк) 2003; (2). Available from: http://www.1796kotok.com/vaccines/malady/noreiko.htm.
Библиографическое описание: Супотницкий М.В. «Забытая» иммунология эпидемических, инфекционных и поствакцинальных процессов // Новости медицины и фармации. — 2014. — № 9–10. — С. 19–23; № 11–12. — С. 16–20.
ССЫЛКИ ПО ТЕМЕ
[1] Ранее нами были рассмотрены подходы к обнаружению обоих феноменов при доклиническом изучении иммунобиологических лекарственных препаратов (ИЛП) [8–10].
[2] Т-хелперы 1 (Th1) – преимущественно способствуют развитию клеточного иммунного ответа, активируя Т-киллеры; Т-хелперы 2 (Th2) – активируют В-лимфоциты, способствуя развитию гуморального иммунного ответа.
[3] Этот феномен еще называют правилом Бернета (Burnet's rule) [5].
[4] В западной научной литературе популярен термин «феномен первичного антигенного греха» («phenomenon of original antigenic sin», OAS). Для его образования Thomas Francis, Jr. (1900–1969) [13] использовал библейское выражение «original sin», означающее «первородный (прародительский, первичный) грех» Адама, отразившийся на всех его потомках. Из-за произвольных и часто несерьезных толкований термина, связанных с его библейским происхождением, в данной работе он не используется. В советской научной литературе 1960-х гг. для описания данного феномена использовался термин «анамнестический ответ» («anamnestic response»), см., например, работу Горбуновой [14]. В более поздней работе Ma et al. [15] используется термин «антигенный импринтинг» («antigenic imprinting»), т.е. «антигенный отпечаток».
[5] H (гемагглютинин) – поверхностный белок вируса гриппа, обеспечивающий способность вируса присоединяться к клетке-хозяину. Основные иммуногенные детерминанты располагаются на поверхности вириона в структуре гемагглютинина, индуцируют в организме образование нейтрализующих антител и кодируются 4-м сегментом вирионной РНК вируса гриппа. Гемагглютинин вируса гриппа представляет собой тример, построенный из двух различных по структуре участков: трехнитчатой закрученной в спираль конструкции из альфа-спиралей, отстоящей на 7,6 нм от мембраны, и глобулярного участка антипаралельной бета-поверхности,которая содержит сайт связывания рецептора. Антитела к гемагглютинину обеспечивают основной иммунитет против вируса.
N (нейраминидаза) — фермент, относящийся к гликозил-гидролазам. Один из антигенов вируса гриппа. Активность нейраминидазы помогает вирусным частицам проникать через секреты слизистых оболочек, богатых сиаловой кислотой, для достижения вирионами клеток-мишеней эпителия дыхательных путей. Облегчает высвобождение вновь образованных вирусных частиц с поверхности зараженных клеток.
[6] Два варианта гемагглютинина, которые ранее считались подтипами Н0 и Hsw1, сейчас признают вариантами подтипа Н1.
[7] Причины развития пандемии «испанки» никого из тех, кто тогда стоял за массовыми вакцинациями не интересовали в принципе, не интересуют они и нынешних борцов с гриппом. В начале прошедшего десятилетия понятие о научной репутации утонуло в птичьем навозе — в медийном пространстве появился «вирус птичьего гриппа». Однако вирус гриппа, вызывающий эпизоотии среди птиц, по данным молекулярно-биологических исследований, никакого отношения к вирусу гриппа, вызвавшему пандемию 1918 г., не имеет. Но в конце того же десятилетия «видными деятелями науки» стало выгодно топить свою научную репутации в свином навозе. И ее утопили. «Вирус птичьего гриппа» в один (!) из последних апрельских дней 2009 г. заменили «вирусом свиного гриппа», так как тот якобы «мутировал» и опасности уже не представляет. Более подробно об этой научной профанации см. в [92].
[8] P. yoelii yoelii — возбудитель малярии грызунов. Используется для моделирования малярии в экспериментах на мышах
[9] (+)ssРНК означает, что вирус содержит одноцепочечную (single-stranded, ss) плюс-цепь РНК, которая используется им в качестве мРНК и генома.
[10] Авидность, авидитет (лат. aviditas — алчность, страсть) — степень сродства антител к антигену, определяющая скорость и полноту протекания иммунных реакций, а также прочность полученного комплекса «антиген–антитело».
[11] Fc-рецепторы (Fc receptors) — представляют собой семейство молекул, каждый член которого распознает иммуноглобулин одного или нескольких родственных изотипов. Рецепторы этого типа входят в состав суперсемейства иммуноглобулинов. Fc-рецепторы для иммуноглобулинов присутствуют на поверхности мононуклеарных лейкоцитов, нейтрофилов, нормальных клеток-киллеров, эозинофилов, базофилов и тучных клеток. Взаимодействуя с Fc-областью иммуноглобулинов разных изотипов, эти рецепторы стимулируют, например, фагоцитоз, противоопухолевую цитотоксическую активность и дегрануляцию тучных клеток.
[12] Частично раздел по антителозависимому усилению инфекции был опубликован на украинском языке в журнале «Iнфекцiйнi хвороби» [124].
[13] Fc-фрагмент Ig (кристаллизующийся фрагмент иммуноглобулина, fragment crystallizable region, Fc region) — концевая часть молекулы иммуноглобулина, которая взаимодействует с Fc-рецептором на поверхности клетки и с некоторыми белками системы комплемента. Другая часть антитела называется Fab (от англ. Fragment antigen binding), и состоит из двух вариабельных участков, определяющих специфичность мишени, которую связывает антитело. Fc-фрагменты антител одного класса консервативны.
[14] Fc-рецепторы (Fc receptors) — представляют собой семейство молекул, каждый член которого распознает иммуноглобулин одного или нескольких родственных изотипов. Рецепторы этого типа входят в состав суперсемейства иммуноглобулинов. Fc-рецепторы для иммуноглобулинов присутствуют на поверхности мононуклеарных лейкоцитов, нейтрофилов, нормальных клеток-киллеров, эозинофилов, базофилов и тучных клеток. Взаимодействуя с Fc-областью иммуноглобулинов разных изотипов, эти рецепторы стимулируют, например, фагоцитоз, противоопухолевую цитотоксическую активность и дегрануляцию тучных клеток.
[15] Этот процесс отличается от образования иммунных комплексов, приводящих к развитию так называемых иммунокомплексных болезней (ревматоидный артрит, сывороточная болезнь, системная красная волчанка и др.), тем, что комплексы «вирус–специфическое антитело» не вызывают прямого повреждения тканей и органов. Их патологическое действие проявляется через усиление инфекционного процесса. Более подробно с патогенезом иммунокомплексных болезней можно ознакомиться по монографии Н.А. Константиновой [66].
[16] С1 — компонент комплемента. Его субъединица C1q имеет рецептор для связывания с Fc-фрагментом молекулы антитела.
[17] Моноциты образуются в костном мозге из гемопоэтических стволовых клеток-предшественников. Они циркулируют в кровяном русле 1–3 сут, затем созревают в резидентные макрофаги и дендритные клетки. Моноциты — наиболее активные фагоцитирующие клетки периферической крови.
[18] Toll-подобный рецептор — рецептор системы иммунитета, подобный рецепторному белку Toll плодовой мушки (Drosophila).
[19] Геликазы — ферменты, расплетающие двухцепочечные ДНК или РНК у вирусов, бактерий и эукариот.
[20] Однако в слюнных железах переносчиков DENV — комаров, гетерогенизация вируса возобновляется [97]. Мы объясняем это тем, что у членистоногих хорошо развита «питательная среда» для вируса — фагоцитарная система, однако для гуморальных факторов их иммунной системы характерно отсутствие высокой специфичности, присущей антителам позвоночных (см. в работе [98]).
[21] Подробное описание iADE при лихорадке Денге можно найти в работах [50, 101].
[22] В России такая вакцина не зарегистрирована.
[23] В исследованиях ВИЧ-вакцин на людях есть моральный аспект. Ведь феномен ADE при ВИЧ-инфекции открыт еще в 1988 г. [70], а феномен антигенного импринтинга в 1991 г. [43]. Следовательно, исход этих «экспериментов» 1990-х гг. был ясен. Их опасность для вакцинированных добровольцев тогда же совсем нетрудно было спрогнозировать. Но «разработчики» ВИЧ-вакцин 25 лет обманывали и налогоплательщиков, за счет которых шла их якобы разработка; и людей, согласившихся участвовать в таких экспериментах. К тому же было потеряно время. Вместо того, что бы разрабатывать противоэпидемические мероприятия исходя из реалий эпидемиологии и иммунологии ВИЧ/СПИД-пандемии, эти годы впустую потрачены на ожидание спасительной вакцины. Последствия катастрофичны. Если в России в 1994 г. было зарегистрировано 887 случаев ВИЧ-инфекции, то в конце 2013 г. ВИЧ обнаружен уже у 799 тыс. россиян. Нет никаких признаков прекращения ВИЧ/СПИД-пандемии. Каких-либо идей, кроме «создадим вакцину и покончим со СПИДом, как с натуральной оспой» (см. [4]), то же нет. Дальше то что?

Михаил Васильевич Супотницкий
Российский микробиолог, полковник медицинской службы запаса, изобретатель, автор книг и статей по истории эпидемий чумы и других особо опасных инфекций, истории разработки и применения химического и биологического оружия. Заместитель главного редактора научно-практического журнала «Вестник войск РХБ защиты» Министерства обороны РФ.