Феномен антитело-зависимого усиления инфекции у вакцинированных и переболевших

Суть феномена антитело-зависимого усиления инфекции (antibody-dependent enhancement, ADE) состоит в усиление инфекционного процесса в присутствии антител, специфических к возбудителю инфекционной болезни. ADE развивается в две стадии: внешнее ADE (extrinsic ADE, eADE) – вирусспецифическое антитело, образовавшее комплекс с вирусом, посредством взаимодействия его Fc-фрагментас рецептором Fc (FcR) и/или с рецепторами комплемента на поверхности фагоцитирующих клеток, усиливают распространение вируса по фагоцитирующим клеткам; и внутреннее ADE (intrinsic ADE, iADE) – комплексы «вирус-специфическое антитело», взаимодействующие с фагоцитирующей клеткой через Fc-рецепторы и рецепторы комплемента, запускают сигнальные механизмы, блокирующие ее антивирусную защиту и тем самым способствуют внутриклеточному размножению вируса. Феномен ADE развивается при инфекционных процессах, и в ответ на вакцинацию и введение иммуноглобулинов. У ранее вакцинированного человека он может быть связан с: 1) неполноценной иммунизацией; 2) особенностями взаимодействия возбудителя инфекционной болезни с иммунной системой человека. Наиболее вероятно развитие ADE у лиц, ранее вакцинированных в отношении вирусов, возбудителей инфекционных болезней, представителей семейств Orthomyxoviridae, Paramyxoviridae, Rhabdoviridae, Coronaviridae, Retroviridae, Parvoviridae, Filoviridae, Flaviviridae, Togaviridae, Picornaviridae, а также возбудителей туберкулеза. Предложен алгоритм доклинических исследований, имеющих целью обнаружение ADE и установление его природы.
Миронов А.Н., Супотницкий М.В. Лебединская Е. В. Феномен антитело-зависимого усиления инфекции у вакцинированных и переболевших // Биопрепараты. 2013. № 3. С.12–25.
The essence of the antibody-dependent enhancement (ADE) of infection phenomenon is that the infectious process increases in the presence of antibodies, specific to the infectious agents. ADE has two stages of development: external ADE (extrinsic ADE, eADE) – a virus-specific antibody forms an antibody-virus complex, by it's Fc-fragment interaction with Fc-receptor-Fc (FcR) and/or with complement's receptors on the surface of phagocytic cells, it increases viral shedding by phagocytic cells, and inner ADE (intrinsic ADE, iADE) – "virus-specific antibody" complexes which interact with phagocytic cells via Fc-receptors and complement's receptors, triggering signaling mechanisms which block it's antivirus protection and thereby promote the intracellular virus replication. The phenomenon of ADE develops under the condition of infectious processes and as a response to vaccination and immunoglobulin administration. In previously vaccinated person it may be related to: 1) inadequate immunization, 2) features of interaction between the infectious agent and human immune system. The data provided in the article shows that the immunity is not always resistance. Not all the immune reactions to vaccination or an infectious process are defensive. Therefore it is necessary to develop methods of research allowing to detect the possibility of ADE to vaccination or administration of specific immunoglobulins during the pre-clinical studies.
Mironov A.N., Supotnitskiy М.V., Lebedinskaya E.V. The phenomenon of antibody-dependent enhancement of infection in vaccinated and convalescents // Biopreparats (Biopharmaceuticals). 2013. No. 3. P. 12–25.
В последнее десятилетие в научных журналах появилось большое количество публикаций об участии иммунной системы человека в патогенезе инфекционных болезней [30, 31, 56, 68, 77, 79]. Провалом закончились масштабные клинические испытания ВИЧ-вакцины, разрабатываемой фармацевтическим гигантом Merck KGaA в полном соответствии с общепринятыми представлениями о роли Т- и В-систем иммунитета в блокировании инфекционных процессов [60, 63]. Эти и другие данные свидетельствуют о наличии обширных пробелов в знаниях о тонких механизмах функционирования иммунной системы человека, что не может не сказываться на эффективности иммунобиологических лекарственных препаратов (ИЛП) и результативности их экспертизы. Цель работы – обобщить имеющиеся в научной литературе данные по феномену антитело-зависимого усиления инфекции у вакцинированных и переболевших.
Формирование типовых представлений об иммунитете. Теория опсонинов, разработанная А.Е. Райтом и С. Дугласом в 1903 г. [8], примирила враждующие между собой фагоцитарную (И.И. Мечников) и гуморальную (Р. Кох, П. Эрлих) теории иммунитета, и дала толчок к ее дальнейшему развитию на основе представлений о кооперации клеточно-гуморальных иммунологических реакций. В последующие годы были описаны и апробированы иммунологические реакции и тесты с фагоцитирующими клетками и специфическими антителами, уточнялся механизм их взаимодействия с антигенами. В 1948 г. А. Фагреус доказала, что антитела синтезируются плазмоцитами. Иммунологическая роль Т- и В-лимфоцитов установлена в 1960-х гг., когда было показано, что под влиянием антигенов В-клетки превращаются в плазмоциты, а из недифференцированных Т-клеток возникает несколько субпопуляций, синтезирующих специфические к антигену антитела. В 1966 г. открыты цитокины Т-лимфоцитов, обуславливающие кооперацию (взаимодействие) иммункомпетентных клеток. Сформулированная в 1964 г. Н. Йерне и Ф. Бернетом клонально-селекционная теория иммунитета дала ученым понимание того, каким образом специфические антитела могут накапливаться в достаточно высокой концентрации, чтобы эффективно блокировать инфекционный процесс. Подобный же механизм установлен и при формировании клоноспецифических Т-клеток [7].
Сложившаяся в результате этих исследований типовая схема иммунного ответа выглядит следующим образом. Макрофаг поглощает (фагоцитирует) патогенный микроорганизм (бактерия, вирус), инактивирует его и презентируют его Т- и В-лимфоцитам. Ввиду различий рецепторного аппарата В-клетки реагируют с одними детерминантами, Т-клетки – с другими. Получив информацию об антигене от антигенпрезентирующих клеток (макрофаги, несущие на внешней мембране антигены), Т-хэлперы с помощью иммуноцитокинов передают сигнал, усиливающий пролиферацию Т- и В-лимфоцитов определенных клонов[1]. В-лимфоциты дифференцируются до плазмоцитов, а Т-хелперы превращаются в Т-киллеры (Т-эффекторы). Плазмоциты синтезируют специфические антитела, участвующие в иммунном ответе в трех формах: нейтрализации, опсонизации и активации системы комплемента (гуморальный иммунный ответ); Т-киллеры разрушают клетки-мишени при непосредственном контакте (цитотоксический или клеточный иммунный ответ). После первичного контакта с антигеном остаются клоны Т- и В-клеток памяти, сохраняющие информацию о нем много лет. При вторичном попадании этого антигена в организм человека происходит стимуляция клонов, и они начинают быстро размножаться. В-клетки переходят в плазмоциты, продуцирующие антитела нужной специфичности. Т-клетки обеспечивают клеточную форму защиты (субпопуляции цитотоксических Т-клеток и Т-клеток воспаления) и участвуют в формировании гуморального иммунитета – хелперные Т-клетки [1, 2, 7].
В соответствие с этой схемой иммунитет – это невосприимчивость. Все гуморальные и клеточные реакции, развивающиеся в ответ на введение вакцины или развитие инфекционного процесса, имеют защитный характер.
Феномен антителозависимого усиления инфекции (antibody-dependent enhancement, ADE). Феномен ADE впервые описан R. A. Hawkes в 1964 г. [34], обнаружившим повышение продукции различных флавивирусов (японского энцефалита, энцефалита долины Мюррей и др.) в клетках куриного эмбриона, впервые экспонированных к вирусам, находящимся в среде с низким содержанием специфических антител. В последствие он привел доказательства, что увеличение «выхода» вируса в подобных экспериментах вызвано образованием комплекса «вирус–антитело» [35]. Эти данные настолько расходились с общепринятыми представлениями о защитной роли антител в инфекционном процессе, что их посчитали артефактами. Однако в конце 1960-х и начале 1970-х гг. уже другими исследователями обнаружена роль ADE в патогенезе тяжелых форм геморрагической лихорадки, вызванной вирусом Денге (dengue virus, DENV). Было установлено, что наличие антител в сыворотке крови реконвалесцента, оставшихся после легко перенесенных случаев лихорадки Денге, приводит к тяжелому течению болезни, если произошло повторное заражение DENV другого серотипа [30, 31].
Систематически феномен ADE изучается с конца 1980-х гг. [72]. Суть феномена ADE состоит в усиление инфекционного процесса в присутствии антител, специфических к возбудителю инфекционной болезни. ADE развивается в две стадии:
внешнее ADE (extrinsic ADE, eADE) – вирусспецифическое антитело, образовавшее комплекс с вирусом, посредством взаимодействия его Fc-фрагмента[2] с рецептором Fc (FcR)[3] и/или с рецепторами комплемента на поверхности фагоцитирующих клеток, усиливает распространение вируса по фагоцитирующим клеткам;
внутреннее ADE (intrinsic ADE, iADE) – комплексы «вирус–специфическое антитело»[4], взаимодействующие с фагоцитирующей клеткой через Fc-рецепторы и рецепторы комплемента, запускают сигнальные механизмы, блокирующие ее антивирусную защиту и тем самым способствуют внутриклеточному размножению вируса.
В основном феномен ADE проявляется в ответ на образование антител изотипа IgG1 [37]. У людей имеются три типа рецепторов Fc, которые связывают IgG: это сиалогликопротеины FcγRI, FcγRII и FcγRIII (CD16). FcγRI наиболее представлен на моноцитах/макрофагах человека, и он связывает IgG с наибольшей авидностью. Поэтому ему принадлежит «лидерство» среди других рецепторов макрофагов в реализации феномена ADE. ADE, показанный в условиях in vitro, не обязательно воспроизводится в условиях in vivo [45].
Феномен ADE характерен для инфекционных процессов, вызываемых вирусами, имеющими следующие особенности: а) обычно они реплицируются в макрофагах; б) индуцируют продукцию большого количества антител с низкой способностью к нейтрализации гомологичных вирусов; в) способны к персистентной инфекции, характеризующейся продолжительной виремией [73]. Феномен ADE также обнаружен при инфекционных процессах, вызываемых бактериальными патогенами, но изучен фрагментарно. Например, порообразующий токсин золотистого стафилококка – лейкоцидин, усиливает свое токсическое действие, если в крови человека содержатся специфические к нему антитела [85]. Такой же эффект вызывают моноклональные антитела к токсину А патогенных клостридий [36]. Имеются косвенные доказательства причастности феномена ADE к прогрессированию туберкулезной инфекции и Ку-лихорадки. При аэрозольном инфицировании M. tuberculosis мышей 57BL/6, дефицитных по рецептору FcγIIB, патологические изменения у них развиваются через 30 сут, у интактных мышей через 20 сут [49]. В условиях in vitro показано, что антитела к C. burnetii I фазы стимулируют ее размножение в макрофагах более эффективно, чем антитела к этому же микроорганизму II фазы [61, 65].
Возможно, что первое описание ADE оставил Д.К. Заболотный, наблюдавший в 1899 г. в Вэнчане (Монголия) появление пустулезной формы чумы у больного с бубонной чумой на пятые сутки после введения противочумной сыворотки. Он объяснил это явление примерно так, как сегодня объясняют ADE – антитела к возбудителю чумы распространили его по фагоцитирующим клеткам и усилили инфекционный процесс [3]. Можно предположить, что из-за низкого качества противочумной сыворотки, примененной Д.К. Заболотным, и ненадлежащих условий ее хранения во время экспедиции к очагам чумы в Монголии, антитела к возбудителю чумы утратили нейтрализующее действие, но сохранили способность взаимодействовать с FcR.
eADE. Феномен наблюдается в двух вариантах: а) комплемент-опосредованное антителозависимое усиление инфекции (complement-mediated ADE; C-ADE); и б) независящее от комплемента и связанное с Fc-рецептором усиление инфекции (Fc-receptor-mediated ADE; FcR-ADE) [68, 73] (рисунок 1).
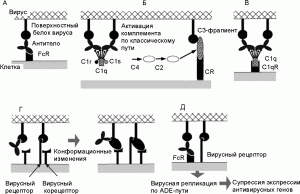 Рис. 1. Общая схема развития феномена eADE при вирусных инфекциях. А. Взаимодействие между антителом и FcR на поверхности макрофага. Б. Фрагмент С3 комплемента (компонент комплемента, после присоединения которого весь этот комплекс приобретает способность прилипать к различным частицам и клеткам) и рецептор комплемента (complement receptor; CR) способствуют присоединению вируса к клетке. В. Белки комплемента С1q и С1qR способствуют присоединению вируса к клетке (в составе молекулы C1q имеется рецептор для связывания с Fc-фрагментом молекулы антитела). Г. Антитела взаимодействуют с рецептор-связывающим сайтом вирусного белка и индуцируют его конформационные изменения, облегчающие слияние вируса с мембраной. Д. Вирусы, получившие возможность реплицироваться в данной клетке посредством eADE, супрессируют антивирусные ответы со стороны антивирусных генов клетки. По[68]
Рис. 1. Общая схема развития феномена eADE при вирусных инфекциях. А. Взаимодействие между антителом и FcR на поверхности макрофага. Б. Фрагмент С3 комплемента (компонент комплемента, после присоединения которого весь этот комплекс приобретает способность прилипать к различным частицам и клеткам) и рецептор комплемента (complement receptor; CR) способствуют присоединению вируса к клетке. В. Белки комплемента С1q и С1qR способствуют присоединению вируса к клетке (в составе молекулы C1q имеется рецептор для связывания с Fc-фрагментом молекулы антитела). Г. Антитела взаимодействуют с рецептор-связывающим сайтом вирусного белка и индуцируют его конформационные изменения, облегчающие слияние вируса с мембраной. Д. Вирусы, получившие возможность реплицироваться в данной клетке посредством eADE, супрессируют антивирусные ответы со стороны антивирусных генов клетки. По[68]
В таблице 1 обобщены сведения по вирусным и бактериальным инфекциям, сопровождающимся феноменом еADE.
Таблица 1. Инфекционные болезни, сопровождающиеся феноменом еADE*
|
Инфекционная болезнь |
Возбудитель болезни (семейство) |
Тип ADE |
Примечание |
Источник |
|
Вирусные инфекции |
||||
|
ВИЧ/СПИД |
Human immunodeficiency virus, HIV, ВИЧ (Retroviridae) |
FcR-ADE, C-ADE, |
На ранней стадии инфекции феномен реализуется через V3-петлю gp120 (по типу FcR-ADE). По типу C-ADE феномен начинает проявляться перед клиническим прогрессированием ВИЧ-инфекции |
[38, 62, 72] |
|
Инфекционная анемия лошадей |
Equine infectious anemia virus, EIAV (Retroviridae) |
FcR-ADE |
Показано обострение болезни у вакцинированных лошадей и пони, как следствие присутствия антител, индуцированных введением инактивированной цельновирионной вакцины или рекомбинантной вакцины, полученной на основе поверхностного гликопротеина EIAV |
[42, 81]
|
|
Иммунодефицит кошек |
Feline immunodeficiency virus, FIV (Retroviridae) |
FcR-ADE |
Вакцинирование оболочечным рекомбинантным белком вируса усиливает виремию. Усиление клинических признаков болезни у кошек при росте титров антител к коровому белку (core protein) FIV |
[39, 59, 70]
|
|
Инфекционный перитонит кошек |
Feline infectious peritonitis virus, FIPV, (Coronaviridae) |
FcR-ADE |
Феномен ADE проявляется при инфицировании вирусом того же серотипа, в отношении которого был иммунный ответ |
[39] |
|
Лихорадка Эбола |
Вирус Эбола (Filoviridae) |
FcR-ADE, C-ADE |
Сыворотка, взятая от мышей, иммунизированных ДНК вакциной с геном поверхностного гликопротеина вируса Zaire, вызывает ADE |
[69] |
|
Лихорадка Марбург |
Вирус Марбург (Filoviridae) |
C-ADE |
ADE наиболее выражен при инфекции, вызванной вирулентный изолятом Angola |
[56] |
|
Гепатит С |
Вирус гепатита С (Flaviviridae) |
FcR-ADE |
ADE рассматривается как основная причина, мешающая созданию эффективных вакцин |
[58] |
|
Желтая лихорадка |
Viscerophilus tropicus (Flaviviridae) |
FcR-ADE |
ADE рассматривается как основная причина нейровирулентности вируса и тяжелого течения болезни. Показана связь ADE с антителами к гликопротеину Е вируса |
[14, 29]
|
|
Корь |
Measles virus (Paramyxoviridae) |
FcR-ADE |
ADE вызывают антитела к гемагглютинину Н. Авторы связывают с ADE случаи так называемой атипичной кори, когда она развивается у ранее вакцинированных людей и протекает в тяжелой форме |
[41] |
|
Энцефалит Западного Нила |
West Nile virus, WNV (Flaviviridae) |
C-ADE |
ADE вызывают субклассы IgG, с высокой афинностью к фактору комплемента C1q |
[53] |
|
Энцефалит долины Мюррей |
Murray Valley encephalitis virus, MVEV (Flaviviridae) |
Нет данных |
Антитела к вирусу японского энцефалита в субнейтрализующих концентрациях увеличивают вирусемию и смертность среди мышей, зараженных MVEV. Авторы выражают опасение, что программы по вакцинации населения к вирусу японского энцефалита в тех районах, где одновременно с ним циркулирует и MVEV, могут способствовать развитию эпидемии энцефалита долины Мюррей |
[80] |
|
Лихорадка Денге |
Dengue virus, DENV, DV (Togaviridae) |
FcR-ADE |
Геморрагическая лихорадка развивается при перекрестном инфицировании любым из серотипов вируса |
[12]
|
|
Энтероинфекции и патология ЦНС |
Human enterovirus 71, EV71 (Picornaviridae) |
FcR-ADE |
Корреляция между ADE и тяжелым течением болезни |
[33] |
|
Бешенство |
Вирус бешенства (Rhabdoviridae) |
Нет данных |
Антитела к вирусу усиливают инфицирование макрофагов в условиях in vitro через образование иммунных комплексов. Предполагается связь этого феномена с «ранней смертью» от бешенства у вакцинированных животных |
[15, 44, 58] |
|
Алеутская болезнь норок |
Aleutian disease virus, ADV (Parvoviridae) |
FcR-ADE |
Комплекс «ADV–антитело» осаждается на ренальных гломерулярных мембранах или стенках капиллярных сосудов почек, вызывая летальный гломерулонефрит |
[57]
|
|
Бактериальные инфекции |
||||
|
Стафилококковая инфекция |
Лейкоцидин Пантон-Валентина (Panton-Valentine leukocidin, PVL) – пороформирующий цитотоксин золотистого стафилококка (Micrococcaceae) |
Нет данных |
Антитела к PVL, присутствующие в крови большинства людей, усиливают действие PVL, повышая патогенность PVL-продуцирующих S. aureus |
[85] |
|
Клостридиоз |
Токсин A, toxin TcdA Clostridium difficile (Clostridiaceae) |
FcR-ADE |
Моноклональные антитела к токсину А усиливают его поражающее действие |
[36] |
|
Туберкулез |
M. tuberculosis (Mycobacterium tuberculosis complex) |
FcR-ADE (предпо-ложи-тельно) |
У мышей 57BL/6, дефицитных по рецептору FcγIIB, при аэрозольном инфицировании M. tuberculosis патологические изменения развиваются медленнее, чем у интактных мышей |
[49] |
|
Лихорадка Ку |
Coxiella burnetii (Rickettsiella) |
Нет данных |
Антитела к C. burnetii I фазы стимулируют ее размножение в макрофагах |
[61]
|
* За основу взята таблица, опубликованная нами ранее [10].
Безоболочечным вирусам (non-enveloped viruses), образовавшим комплекс с антителом, способным взаимодействовать с Fc-рецептором, специфические рецепторы на поверхности клетки-мишени не требуются [68].
Компонент комплемента С1[5], связывая Fc-фрагмент антитела, инициирует классический путь активации комплемента, в результате чего активируется компонент комплемента С3, ковалентно (!) связывающийся или с антителом или с поверхностью вирусной частицы. Образовавшийся комплекс способен взаимодействовать с рецепторами комплемента на поверхности клетки посредством С3, усиливая взаимодействие вируса с клеткой. Альтернативно C1q-субъединица непосредственно может перекрестно связывать вирусный белок и C1q-рецепторы (C1qR) на поверхности фагоцитирующих клеток. Все перечисленные эффекты находятся в зависимости от концентрации антител.
iADE. Толчком к исследованиям блокирующего действия ADE на антивирусную защиту клетки послужили данные, полученные при изучении причин развития хронических артритов у реконвалесцентов, перенесших острую форму болезни, вызванную вирусом Росс Ривер (Ross River virus, RRV). Такие артриты могут длиться до года, делая пациента на весь этот период неработоспособным. В синовиальной жидкости пациентов с хроническими артритами обнаружены антигены RRV и γ-интерферон (IFN-γ), что свидетельствует о хронической RRV-инфекции. При попытке ее воспроизвести на линиях мышиных макрофагов и первичных человеческих моноцитов/макрофагов[6] (primary human monocytes/macrophages) установлено, что инкубирование RRV с разбавленной специфической сывороткой приводит: 1) к супрессии синтазы оксида азота 2 (nitric oxide synthase 2, NOS2) и, соответственно, к снижению продукции активных радикалов азота (reactive nitrogen radical); 2) прекращению экспрессии генов интерферон-регулирующего фактора 1 (interferon regulatory factor 1, IRF-1) и фактора ядра каппа-би (nuclear factor-κВ) и, соответственно, к подавлению синтеза фактора некроза опухолей альфа (tumor necrosis factor alpha, TNF-alpha) и IFN-γ; 3) заметному увеличению синтеза интерлейкина-10 (interleukin-10, IL-10). Лигирование FcγR с комплексом антитело-зимозан в присутствии RRV не вызывало вышеописанного эффекта [47, 48]. Следовательно, увеличение продукции вируса клетками при ADE вызвано не только увеличением возможностей для взаимодействия вируса с поверхностью макрофагов, но и подавлением их собственной системы защиты от вирусов (innate cellular immunity) [76].
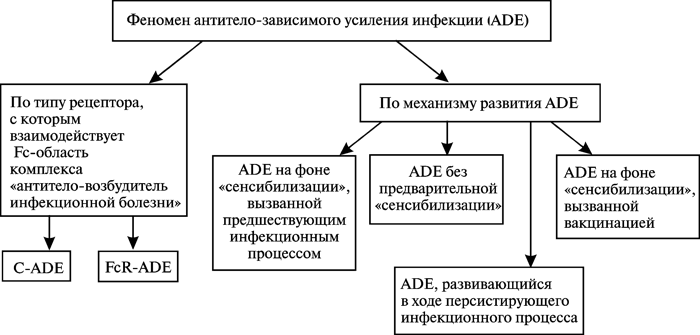
Классификация феномена ADE. Приведенные данные позволяют нам предложить классификацию феноменов ADE по двум принципам: по типу рецептора, с которым вирус взаимодействует на поверхности моноцитов/макрофагов (С-ADE и FcR-ADE), и по механизмам развития ADE (рисунок 2).
Первая классификация удобна для изучения феномена ADE в условиях in vitro, например, для установления границ феномена среди близкородственных видов вирусов на клетках культур тканей, содержащих, либо наоборот, не содержащих Fc- и CIq-рецепторы; либо при их блокировании специфическими мАТ (см. таблицу 1). Границы феномена ADE устанавливаются с помощью специфических сывороток к вирусам близкородственных видов. Вторая классификация – для воспроизведения ADE в условиях in vivo при разработке ИЛП и их доклинических и клинических исследованиях.
Феномен ADE, развивающийся на фоне «сенсибилизации», вызванной предшествующим инфекционным процессом. Наиболее изучен среди других проявлений феномена ADE, поэтому мы рассмотрим его более подробно, чем остальные. Опережающим объектом исследований при изучении феномена данного типа является геморрагическая лихорадка Денге – острая трансмиссивная инфекция, распространенная в странах Южной и Юго-Восточной Азии, Африки, Океании и Карибского бассейна.
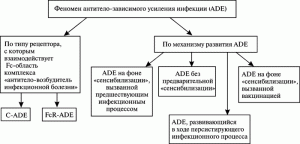 Рис. 2. Классификация ADE.
Рис. 2. Классификация ADE.
Возбудитель лихорадки Денге – оболочечный (+)ssРНК-вирус[7], четыре серотипа которого (DENV1–DENV4) относятся к арбовирусам семейства Togaviridae рода Flavivirus (арбовирусы антигенной группы В). Передача возбудителя инфекции среди людей осуществляется комарами Aedes aegypti, среди обезьян – A. albopictus. Клинически болезнь характеризуется развитием геморрагического диатеза и тенденцией к развитию шокового состояния (шоковый синдром Денге), который может привести к смерти. Отдельные вспышки болезни могут охватывать сотни тысяч человек. Ежегодно в мире не менее 50 млн человек заболевают лихорадкой Денге [25]. Схема жизненного цикла DENV в отсутствие специфических антител представлена на рисунке 3.
После проникновения DENV в эндосомы, клетка запускает механизмы антивирусной защиты [74], в частности, экспрессию интерферонов (IFN). Оба типа интерферонов – тип I (α, β) и тип II (γ) способны блокировать репликацию DENV, если происходит его распознавание эндосомальными рецепторами: toll-подобный рецептор 3 (toll-like receptor, TLR-3)[8] – распознает двухцепочечную РНК (dsRNA) вируса; TLR8 – распознает G-богатые олигонуклеотиды; и TLR7 – распознает ssРНК.
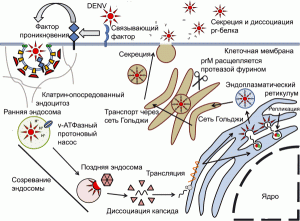 Рис. 3. Жизненный цикл DENV в отсутствие специфических антител. Вирус проникает в клетку по механизму рецептор-опосредованного эндоцитоза. Клеточная мембрана впячивается внутрь клетки, формируя окаймленные ямки. Внутриклеточная сторона окаймленной ямки в основном включает белок клатрин (clathrin). В зависимости от серотипа вируса или типа клетки, вирус при рН 6,0 сливается со стенкой ранней эндосомы и покидает ее, либо это происходит при рН 5,0–6,0 уже в поздней эндосоме. В эндоплазматическом ретикулуме (endoplasmic reticulum, ER) (+)РНК транслируется на рибосомах с образованием отдельного полипротеина, который в последующем процессируется с помощью аутопротеаз и клеточных протеаз на 7 неструктурных белков (NS1, 2A, 2B, 3, 4A, 4B и 5) и три структурных белка: C (капсид), prM (прекурсорный мембранный белок, precursor membrane protein) и E-белок. Неструктурные белки инициируют репликацию РНК вируса, prM и E-белки формируют гетеродипмеры, которые ориентированы в просвет эндоплазматического ретикулума, где происходит частичная сборка вирионов. РНК вируса ассоциируется с С-белком и формирует нуклеокапсид, который «одевается» липидной мембраной, содержащей гетеродимеры prM- и Е-белков. Из цитоплазмы клетки вирионы выводятся через сеть Гольджи, где происходит их созревание – prM расщепляется сериновой протеазой фурином с образованием растворимого pr-белка и М-белка. pr-белок остается ассоциированным с Е-белком во время экзоцитоза, предотвращая преждевременное слияние вирионов с участками сети Гольджи, имеющими кислые значения рН. Попав в нейтральную среду межклеточного пространства pr-белок диссоциирует, и вирусная частица становится способной вызывать инфекционный процесс, т.е. созревшей. По [25]
Рис. 3. Жизненный цикл DENV в отсутствие специфических антител. Вирус проникает в клетку по механизму рецептор-опосредованного эндоцитоза. Клеточная мембрана впячивается внутрь клетки, формируя окаймленные ямки. Внутриклеточная сторона окаймленной ямки в основном включает белок клатрин (clathrin). В зависимости от серотипа вируса или типа клетки, вирус при рН 6,0 сливается со стенкой ранней эндосомы и покидает ее, либо это происходит при рН 5,0–6,0 уже в поздней эндосоме. В эндоплазматическом ретикулуме (endoplasmic reticulum, ER) (+)РНК транслируется на рибосомах с образованием отдельного полипротеина, который в последующем процессируется с помощью аутопротеаз и клеточных протеаз на 7 неструктурных белков (NS1, 2A, 2B, 3, 4A, 4B и 5) и три структурных белка: C (капсид), prM (прекурсорный мембранный белок, precursor membrane protein) и E-белок. Неструктурные белки инициируют репликацию РНК вируса, prM и E-белки формируют гетеродипмеры, которые ориентированы в просвет эндоплазматического ретикулума, где происходит частичная сборка вирионов. РНК вируса ассоциируется с С-белком и формирует нуклеокапсид, который «одевается» липидной мембраной, содержащей гетеродимеры prM- и Е-белков. Из цитоплазмы клетки вирионы выводятся через сеть Гольджи, где происходит их созревание – prM расщепляется сериновой протеазой фурином с образованием растворимого pr-белка и М-белка. pr-белок остается ассоциированным с Е-белком во время экзоцитоза, предотвращая преждевременное слияние вирионов с участками сети Гольджи, имеющими кислые значения рН. Попав в нейтральную среду межклеточного пространства pr-белок диссоциирует, и вирусная частица становится способной вызывать инфекционный процесс, т.е. созревшей. По [25]
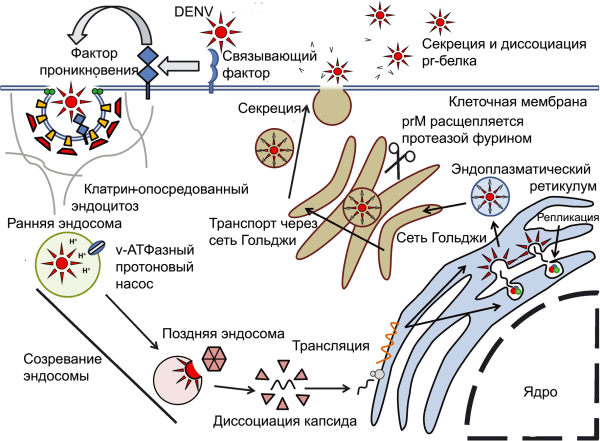
В цитоплазме вирусную РНК «узнают» цитоплазматические РНК-геликазы (cytoplasmic RNA helicases)[9], RIGI (retinoic-acid inducible gene 1) и MDA5 (melanoma differentiation-associated gene 5). Активация TLR индуцирует экспрессию провоспалительных цитокинов: IL8, IL12, IFN-α и IFN-γ. Положительная регуляция экспрессии IL8, осуществляется через ядерный фактор каппа-би (NF-κB). Экспрессия IFN активирует STAT1 и усиливает экспрессию IRF1 (IFN regulatory factor 1), что приводит к усиленной продукции активных радикалов азота (NO). Комбинированное действие интерферонов и NO вызывает антивирусное состояние у соседних клеток (antiviral state) и ограничивает размножение DENV в инфицированных клетках соответственно [75].
При первичном инфицировании человека DENV иммунные ответы на вирус мало отличаются от тех, что описаны в классической схеме иммунного ответа, приведенной выше. Специфичные в отношении DENV B- и T-клетки формируются приблизительно через 6 сут после инфицирования и полностью контролируют развитие инфекции. Вирион DENV распознается антителами, специфичными к белкам E и prM. Структурная организация этих белков у «созревшего» и «несозревшего» вируса различается. Следовательно, различаются и их специфические эпитопы. Доминирующую роль в нейтрализации вируса играют антитела к белку prM «созревшего» вируса. Нейтрализующая активность специфических к DENV антител проявляется на двух уровнях: 1) блокирование взаимодействия вируса с клеточным рецептором; 2) блокирование слияния вируса с клеточной мембраной вследствие связывания антителами петли слияния белка Е. Антитела к prM «несозревшего» вируса обладают перекрестной активностью к DENV всех серотипов, но их нейтрализующая активность незначительна [22, 50].
Репликация DENV, как и любого другого РНК-вируса, сопровождается большим количеством ошибок. Вызвано это тем, что все молекулы вирусной РНК реплицируют через ассиметричную транскрипцию с одной цепи, исключающую большинство корректирующих механизмов, характерных для репликации ДНК. Поэтому первичный инфекционный процесс при лихорадке Денге сопровождается полиморфизацией DENV и образованием квазиспецифичных производных в пределах его серотипа. Иммунная система реагирует на них выработкой специфических антител [46].
При вторичном инфицировании человека DENV гетерологичного серотипа стимулируются клоны В-клеток памяти, сохраняющие информацию о DENV, инфицировавшем человека первично. Они дифференцируются в плазмоциты, продуцирующие антитела к вирусу (его квазипроизводным), который они запомнили, а не к тому, который вызвал инфекцию. Этот иммунологический феномен называется феноменом первичного антигенного греха или антигенным импринтингом[10].
Усиление инфекционного процесса происходит еще до того, как концентрация антител достигнет порога, необходимого для нейтрализации вируса. Продуцируемые плазмоцитами антитела «узнают» DENV, вызвавший инфекционный процесс, но не нейтрализуют его. Они формируют с вирусом комплекс и связывают его с Fc-рецептором на поверхности макрофагов (феномен FcR-ADE), тем самым усиливая инфекционный процесс. Одновременно происходит гомогенизация популяции DENV, так как на этапе eADE преимущества в инфицировании макрофагов/моноцитов получают лишь те квазипроизводные DENV, в отношении которых плазмоцитами вырабатываются антитела, способные связать их с Fc-рецепторами [25, 46][11] (рисунок 4).
Изменения в клетке, связанные с iADE, начинаются раньше, чем DENV покинет эндосому. Точный механизм развития iADE не установлен. Имеющиеся знания позволили [25] описать его следующим образом.
 Рис. 4. Механизм eADE при геморрагической лихорадке Денге. А. Вторичное инфицирование пациента DENV. На различных стадиях инфекции происходит дивергенция DENV. Уже существующие специфические антитела могут блокировать инфекционный процесс, если они встретились с тем его серотипом, который вызвал первичную инфекцию, или, наоборот, усиливать его через механизм ADE, если возбудитель болезни представлен вирусом другого серотипа. Б. Гипотетический механизм гомогенизации популяции DENV на этапе eADE. По [46]
Рис. 4. Механизм eADE при геморрагической лихорадке Денге. А. Вторичное инфицирование пациента DENV. На различных стадиях инфекции происходит дивергенция DENV. Уже существующие специфические антитела могут блокировать инфекционный процесс, если они встретились с тем его серотипом, который вызвал первичную инфекцию, или, наоборот, усиливать его через механизм ADE, если возбудитель болезни представлен вирусом другого серотипа. Б. Гипотетический механизм гомогенизации популяции DENV на этапе eADE. По [46]

Комплекс «DENV–специфическое антитело» через рецептор Fc запускает негативные регуляторы экспрессии TLR3, TLR4, TLR7 и TLR-сигнальных молекул. В результате слабой экспрессии этих рецепторов вирус, проникший в эндосому, не узнается клеткой, эффективной экспрессии генов, кодирующих интерфероны и синтез противовоспалительных цитокинов IL8, IL12, не происходит. Одновременно блокируется экспрессия IRF1, что тормозит продукцию активных радикалов азота[12]. Подавление системы противовирусной защиты клетки приводит к длительному размножению в них DENV, и к увеличению выхода зрелых вирусных частиц [46]. Однако только персистированием DENV в макрофагах iADE при лихорадке Денге не ограничивается.
По сигнальным путям, инициируемым через рецептор Fc, запускается экспрессия гена IL-10, макрофаг начинает продуцировать большие количества IL-10, ингибирующего синтез противовоспалительных цитокинов (IFN-γ, IL 2, 3, 12 и др.) и усиливающего синтез фактора некроза опухолей (TNF) и IL 6, вызывающих повышенную проницаемость сосудов [86]. IL-10 также нарушает дифференциацию Т-хелперов на субпопуляции Th1 и Th2, что ведет к нарушению взаимодействия между клеточными и гуморальными звеньями иммунной системы при блокировании размножения DENV [19]. Лихорадка Денге развивается в тяжелой клинической форме.
Исследования, проведенные с целью выяснить, какие аминокислотные замены структурных и неструктурных белков различных серотипов DENV (мутации в их генах) ассоциируются с тяжелым течением болезни, не дали результатов. Повышенная виремия и высокие количества IL-10 в сыворотке крови всегда сопровождают тяжелое состояние больного. Других объяснений тяжелых осложнений при геморрагической лихорадке Денге, кроме как вовлечения в патогенез болезни ADE, пока не предложено [46, 76].
Феномен ADE, развивающийся без предварительной «сенсибилизации» иммунной системы. A. Takada et al. [67–69] показали, что ADE при инфекционном процессе, вызванном вирусом Эбола (субтип Zaire), развивается в результате взаимодействия образующихся вирусспецифических антител с вирусом и Fc1-рецептором или компонентом комплемента C1q и его рецептором (C1ADE) у макрофагов. Используя моноклональные антитела, исследователи локализовали такие эпитопы у GP вируса субтипа Zaire, и сконструировали химерные эпитопы, индуцирующие продукцию антител у мышей со сниженной способностью вызывать ADE, но обладающих нейтрализующей активностью в отношении вируса субтипа Zaire. Феномен ADE был менее выражен для не опасного для человека субтипа Reston, чем для вирусов субтипов Zaire и Sudan. Авторы данных работ предположили, что феномен ADE играет важную роль в патогенезе лихорадки Эбола (рисунок 5).
Для лихорадки Марбург феномен ADE был описан в 2011 г. Так же как для субтипов вируса Эбола, показана связь между ADE и вирулентностью изолятов вируса Марбург. Авторами делается вывод, что феномен ADE лежит в основе патогенеза не только лихорадок Марбург и Эбола, но и других филовирусных геморрагических лихорадок [56].
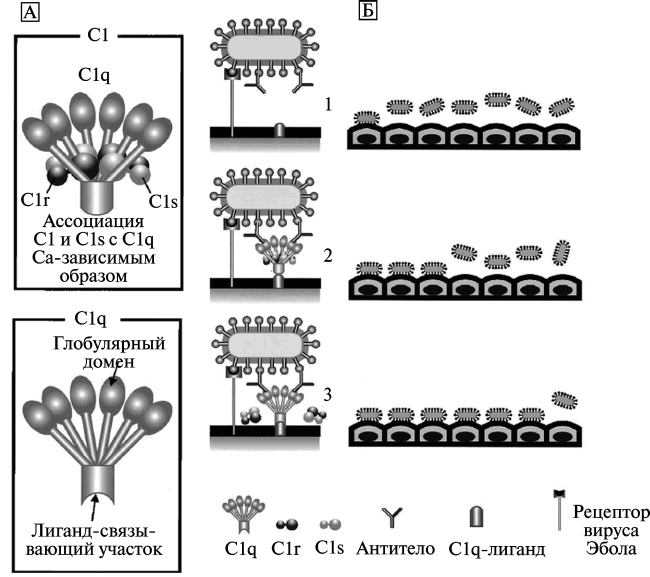
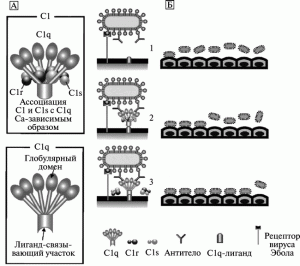 Рис. 5. Модель C-ADE (C1q-ADE) при лихорадке Эбола. A. Схематическое изображение белков комплемента C1 и C1q. Молекула C1q включает глобулярный и лигандсвязывающий домены. Глобулярный домен состоит из шести глобулярных набалдашников (globular heads), которые связываются с Fc1-участком антитела. Лигандсвязывающий домен C1q взаимодействует с лигандом на поверхности фагоцитирующей клетки. Аффинитет C1q к лиганду снижается при ассоциации с C1r и C1s (сериновые протеазы). Б. Механизм ADE при лихорадке Эбола. Вирус Эбола инициирует инфекционный процесс путем связывания со специфическими рецепторами на поверхности фагоцитирующей клетки (1). C1q связывает комплекс «вирус–антитело» с C1q1-лигандами, расположенными на поверхности клеток, вызывая взаимодействие между вирусом и рецептором (2). Диссоциация C1r и C1s от C1q увеличивает связывающий аффинитет молекулы C1q с лигандами на поверхности фагоцитирующей клетки (3). По [68]
Рис. 5. Модель C-ADE (C1q-ADE) при лихорадке Эбола. A. Схематическое изображение белков комплемента C1 и C1q. Молекула C1q включает глобулярный и лигандсвязывающий домены. Глобулярный домен состоит из шести глобулярных набалдашников (globular heads), которые связываются с Fc1-участком антитела. Лигандсвязывающий домен C1q взаимодействует с лигандом на поверхности фагоцитирующей клетки. Аффинитет C1q к лиганду снижается при ассоциации с C1r и C1s (сериновые протеазы). Б. Механизм ADE при лихорадке Эбола. Вирус Эбола инициирует инфекционный процесс путем связывания со специфическими рецепторами на поверхности фагоцитирующей клетки (1). C1q связывает комплекс «вирус–антитело» с C1q1-лигандами, расположенными на поверхности клеток, вызывая взаимодействие между вирусом и рецептором (2). Диссоциация C1r и C1s от C1q увеличивает связывающий аффинитет молекулы C1q с лигандами на поверхности фагоцитирующей клетки (3). По [68]
Феномен ADE, развивающийся в ходе персистирующего инфекционного процесса. Феномен ADE лежит в основе патогенеза болезни многих персистирующих инфекционных процессов. Например, клинически выраженный кошачий инфекционный перитонит, вызываемый FIPV (семейство Coronaviridae), развивается у кошек, уже имевших антитела после ранее перенесенной бессимптомно инфекции, либо на фоне персистирующей инфекции в случае мутации вируса, приведшей к появлению его нового антигенного варианта. Отличить же вирулентные штаммы FIPV от невирулентных в прямых опытах на животных, не удается [78, 79].
Алеутская болезнь норок вызывается парвовирусом (Aleutian disease virus, ADV) из семейства Parvoviridae. ADV патогенен для норок всех цветных вариантов. Основной источник вируса – переболевшие норки-вирусоносители, выделяющие вирус с мочой, калом и слюной. Репликация ADV в макрофагах сопровождается секрецией плазматическими клетками большого количества антител, не обладающих способностью нейтрализовать вирус. Эти антитела образуют иммунные комплексы с ADV, увеличивающие инфицированность макрофагов и вызывающие образование не нейтрализующих антител. «Порочный круг» замыкается осаждением комплекса «ADV–антитело» на ренальных гломерулярных мембранах или стенках капиллярных сосудов почек, что приводит к летальному гломерулонефриту [57].
Но наиболее интересную роль феномен ADE играет при ВИЧ-инфекции. Для ВИЧ он показан в конце 1980-х гг. [38, 62], но до сих пор игнорируется разработчиками ВИЧ-вакцин.
У ВИЧ-инфицированных людей соблюдается определенная очередность проявления вариантов развития eADE. На ранней стадии инфекции феномен реализуется через V3-петлю gp120 (по типу FcR-ADE); по типу C-ADE феномен начинает проявляться перед клиническим прогрессированием ВИЧ-инфекции [72]. Клиническое значение феномена ADE для ВИЧ – это прогрессирование инфекции и облегчение переноса вируса от матери к плоду [28]. Вне контекста представлений о роли ретровирусов в эволюции клеточных форм жизни и роли ADE в эволюции ВИЧ, процесс накопления разных вариантов ВИЧ в популяциях людей выглядит случайным, как проявление некой способности ВИЧ «постоянно меняться». Но случайностей в этом процессе нет.
По данным А. Takeda et al. [71], в условиях in vitro добавление к клеткам моноцитов сыворотки ВИЧ-инфицированных людей в субнейтрализующих концентрациях значительно усиливает репликацию вируса, т.е. на ранних этапах выработки антител к новому серотипу вируса основную роль в усилении инфекционного процесса играет феномен ADE. Высокие концентрации такой сыворотки в условиях in vitro показывают вируснейтрализующую активность. Следовательно, ВИЧ не удается «увильнуть» от специфических антител, однако блокирования инфекционного процесса специфическими антителами в условиях in vivo не происходит. Высокая скорость мутаций при обратной транскрипции и высокая скорость репликации ВИЧ генерируют большое количество серовариантов ВИЧ. Особенно этот процесс дает о себе знать после сероконверсии и перехода болезни в асимптоматическую стадию.
Как только уровень антител, нейтрализующих данный серотип ВИЧ, достигает определенного порога, селекционируется вариант вируса, способный избегать их нейтрализующего действия [17]. Выработка антител к нему начинается заново. И вновь путем вовлечения в инфекционный процесс феномена ADE новому серотипу вируса обеспечивается распространение по клеткам, содержащим на своей поверхности Fc-рецептор (ранняя стадия инфекции) и рецептор комплемента (перед клиническим прогрессированием ВИЧ-инфекции). С каждым новым серовариантом вируса цикл повторяется. Скорость появления как ВИЧ-нейтрализующих антител, так и избегающих их вирусов, варьируют у разных лиц, однако сам цикл многократно повторяется на протяжении жизни ВИЧ-инфицированного человека и больного СПИДом [26], приводя к росту генетического разнообразия ВИЧ. Только по мере истощения иммунной системы и, соответственно, работы маховика ADE, гетерогенизация ВИЧ прекращается. Эту закономерность хорошо иллюстрируют данные R. Shankarappa et al. [64].
У ВИЧ-инфицированных пациентов, так называемых умеренных прогрессоров (moderate progressors), в пределах асимптоматической стадии ВИЧ-инфекции R. Shankarappa et al. [64] выделяют три фазы дивергенции и три фазы роста разнообразия ВИЧ. Под дивергенцией (divergence) эти авторы понимают различия между нуклеотидной последовательностью исходного вируса и последовательностью вируса, полученного от ВИЧ-инфицированного человека через какое-то время после инфицирования. Под разнообразием (diversity) – различия в нуклеотидных последовательностях ВИЧ в данной временной точке (рисунок 6).
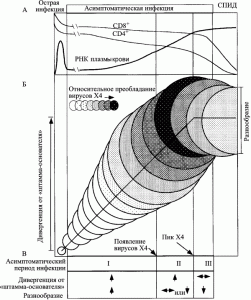 Рис. 6. Схематическое изображение развития ВИЧ-инфекции у умеренных прогрессоров. Диаметры кругов приблизительно соответствуют разнообразию (diversity) вирусной популяции от сероконверсии (первый круг). Вертикальное смещение кругов показывает степень дивергенции вирусной популяции (divergence) от предкового штамма (founder strain). Затенения соответствуют пропорции вирусной популяции, представленной X4-генотипом. Вертикальные линии (начиная с левой стороны схемы) соответствуют: окончанию стадии острой инфекции; пику вирусного разнообразия; стабилизации дивергенции от предкового штамма; развитию СПИДа. В начале поздней фазы дивергенции (роста разнообразия) количество X4-вариантов ВИЧ начинает снижаться. Эта фаза проявляется нарушением гомеостаза Т-клеток. Количество CD4+ T-клеток снижается до уровня <200 клеток/мм3, появляются симптомы выраженного поражения клеточной системы иммунитета, болезнь переходит в стадию СПИДа. Теперь разнообразие вариантов вируса идет на убыль, так как иммунная система истощена и уже не способна раскручивать маховик его эволюции. По [64]
Рис. 6. Схематическое изображение развития ВИЧ-инфекции у умеренных прогрессоров. Диаметры кругов приблизительно соответствуют разнообразию (diversity) вирусной популяции от сероконверсии (первый круг). Вертикальное смещение кругов показывает степень дивергенции вирусной популяции (divergence) от предкового штамма (founder strain). Затенения соответствуют пропорции вирусной популяции, представленной X4-генотипом. Вертикальные линии (начиная с левой стороны схемы) соответствуют: окончанию стадии острой инфекции; пику вирусного разнообразия; стабилизации дивергенции от предкового штамма; развитию СПИДа. В начале поздней фазы дивергенции (роста разнообразия) количество X4-вариантов ВИЧ начинает снижаться. Эта фаза проявляется нарушением гомеостаза Т-клеток. Количество CD4+ T-клеток снижается до уровня <200 клеток/мм3, появляются симптомы выраженного поражения клеточной системы иммунитета, болезнь переходит в стадию СПИДа. Теперь разнообразие вариантов вируса идет на убыль, так как иммунная система истощена и уже не способна раскручивать маховик его эволюции. По [64]

Приведенные R. Shankarappa et al. [64] данные показывают, что в раннюю фазу инфекции развиваются оба процесса; промежуточная фаза характеризуется непрерывным увеличением дивергенции ВИЧ, но стабилизацией или даже снижением его разнообразия; поздняя фаза проявляется снижением темпа или даже стабилизацией процессов дивергенции и формирования разнообразия вируса. Результатом работы такого механизма являются: 1) массивное распространение ВИЧ по фагоцитирующим клеткам; 2) повышение его вирулентности за счет отбора вариантов, тропных к рецептору CXCR4.
По данным Zhang H. et al. [87] увеличение генетического разнообразия вируса субтипа С у детей зависит от антител с широким нейтрализующим действием. Чем выше титр таких антител, тем больше на данный момент времени вирусы различаются между собой.
То, что ВИЧ меняется не сам, а его в ходе инфекционного процесса меняет иммунная система с помощью феномена ADE и специфических антител, выглядит странно только в контексте медицинского подхода к пониманию ВИЧ/СПИД-пандемии. ВИЧ относится к семейству Retroviridae. Вирусы этого семейства интегрируют свою ДНК-копию (провирус) с геномом хозяина в единую молекулу ДНК. Если ретровирус становится частью генома вида, то вид считается прошедшим через эндогенизацию. Эндогенные ретровирусы активны в геноме вида и его видов-потомков до 6 млн лет. Они передаются вертикально, инициируя наращивание его генетического материала образованием своих новых копий; усложняя геном образованием новых экзонов из интронов и/или увеличивая количества генов, подвергающихся альтернативному сплайсингу [9, 10, 13, 20, 21].
Феномен ADE, развивающийся на фоне «сенсибилизации», вызванной вакцинацией. Осложнения после вакцинации, возникающие как следствие феномена ADE, до настоящего времени не стали объектом системных исследований, поэтому сведения о них носят разрозненный характер (таблица 2).
Таблица 2. Феномен ADE, развивающийся как ответ на вакцинацию*
|
Вирус (семейство) |
Доказан в условиях in vitro |
Доказан в условиях in vivo |
Источник |
|
РНК-вирусы |
|||
|
Вирус гриппа А (Orthomyxoviridae) |
+ |
+ |
[86] |
|
Респираторный синтициальный вирус (Paramyxoviridae) |
+ |
+ |
[10] |
|
Вирус кори (Paramyxoviridae) |
+ |
+ |
[29] |
|
Вирус бешенства (Rhabdoviridae) |
+ |
+ |
[48, 62] |
|
Вирус кошачьего инфекционного перитонита (Coronaviridae) |
+ |
+ |
[87] |
|
Вирус свиного репродуктивного и респираторного синдрома (Coronaviridae) |
+ |
+ |
[88] |
|
Вирус обезьяньей геморрагической лихорадки (Coronaviridae) |
+ |
+ |
[77] |
|
ВИЧ (Retroviridae) |
+ |
? |
[16] |
|
Вирус лошадиной инфекционной анемии (Retroviridae) |
+ |
+ |
[56] |
|
Вирус артрита коз (Retroviridae) |
+ |
+ |
[55] |
|
ДНК-вирусы |
|||
|
Вирус алеутской болезни норок (Parvoviridae) |
+ |
+ |
[61] |
*По [30].
Феномен ADE у ранее вакцинированного человека может быть связан с: 1) неполноценной иммунизацией; 2) особенностями взаимодействия возбудителя инфекционной болезни с иммунной системой человека.
Неполноценная иммунизация. Причинно-следственная связь ADE с неполноценной иммунизацией подробно изучена на примерах инактивированной коревой вакцины и инактивированной вакцины против респираторного синтициального вируса (respiratory syncytial virus, RSV) [27, 43]. Обе вакцины получают путем инактивации вирусов формальдегидом. C начала 1960-х гг., т.е. после начала массовых иммунизаций населения против кори вакцинами, инактивированными формалином[13], среди вакцинированных людей отмечаются случаи так называемой атипичной кори (кори, протекающей в тяжелой форме). I. D. Iankov et al. [41] показали, что в основе ее развития лежит феномен FcR-ADE, вызываемый антителами к гемагглютинину вируса (поверхностный белок Н).
Установлено, что антитела, полученные в отношении антигенных белков вирусов кори и RSV, инактивированных формальдегидом, обладают сниженной протективной способностью по сравнению с антителами, полученными в отношении этих же антигенов живых вакцин. Это вызвано тем, что подвергнутые обработке формалином антигенные белки имеют увеличенное количество активных карбонильных групп, что ведет к нарушению третичной структуры эпитопов [23, 55].
ADE как феномен, характерный для взаимодействия возбудителя инфекционной болезни с иммунной системой человека. Если ADE развивается в ходе инфекционного процесса, то есть основание считать, что феномен будет иметь место у вакцинированных людей и животных, если они будут заражены вирусом, против которого их вакцинировали (см. таблицы 1 и 2).
Показательны результаты экспериментов с вакцинами, разрабатываемыми для специфической профилактики ретровирусных инфекций у животных – инфекционной анемии лошадей и имунодефицита кошек. Также они имели цель моделирование стратегий вакцинации против ВИЧ. Хотя эти эксперименты выполнены еще в 1990-х гг., они до сих пор не вызвали интереса у разработчиков ВИЧ-вакцин.
Инфекционная анемия лошадей вызывается вирусом инфекционной анемии лошадей (Equine infectious anemia virus, EIAV). Болезнь носит нециклический характер, проявляется синдромами лихорадки, анорексии, анемии, выздоровления не наступает. Показано серьезное обострение болезни при заражении EIAV вакцинированных лошадей и пони, если в их сыворотке присутствовали антитела, индуцированные введением вакцины. C. Issel at al. [42] использовали виремию как критерий тяжести болезни и продемонстрировали, что вакцинация инактивированной цельновирионной вакциной не может предотвратить развитие виремии и клинических симптомов болезни у животного, которому введен вирулентный штамм вируса. В экспериментах по заражению гетерологичным штаммом вируса животных, вакцинированных высокоочищенным оболочечным гликопротеином вируса, также не удавалось ни предотвратить виремию, ни развитие клинических симптомов болезни. В последующем S. Wang et al. [81] провели масштабные эксперименты на пони и лошадях по оценке защитной эффективности рекомбинантной вакцины, полученной на основе поверхностного гликопротеина EIAV. Результаты экспериментов показали усиление инфекции у всех предварительно вакцинированных животных.
Ретровирус, возбудитель иммунодефицита кошек (Feline immunodeficiency virus, FIV), после инфицирования кошек, вакцинированных оболочечным рекомбинантным белком этого вируса, обнаруживался в их крови даже раньше, чем у не вакцинированных животных [59]. В сходных исследованиях с различными рекомбинантными FIV-вакцинами было установлено, что в крови животных в ответ на вакцинацию обнаруживаются антитела к оболочечному белку (env) FIV, плохо нейтрализующие вирус в условиях in vitro. У вакцинированных животных вирусная нагрузка была значительно большей, чем у не вакцинированных. При росте титров антител к коровому белку (core protein) FIV у кошек имело место усиление клинических признаков болезни [11, 39, 40].
Сходные результаты были получены в экспериментах на людях по изучению протективного эффекта ВИЧ-вакцины, проведенных в Африке фирмой Merck. Из 741 вакцинированного добровольца 24 впоследствии заразились ВИЧ. В другой группе добровольцев, получивших плацебо, 21 из 762 участников также были инфицированы. Эксперимент был досрочно прекращен [77].
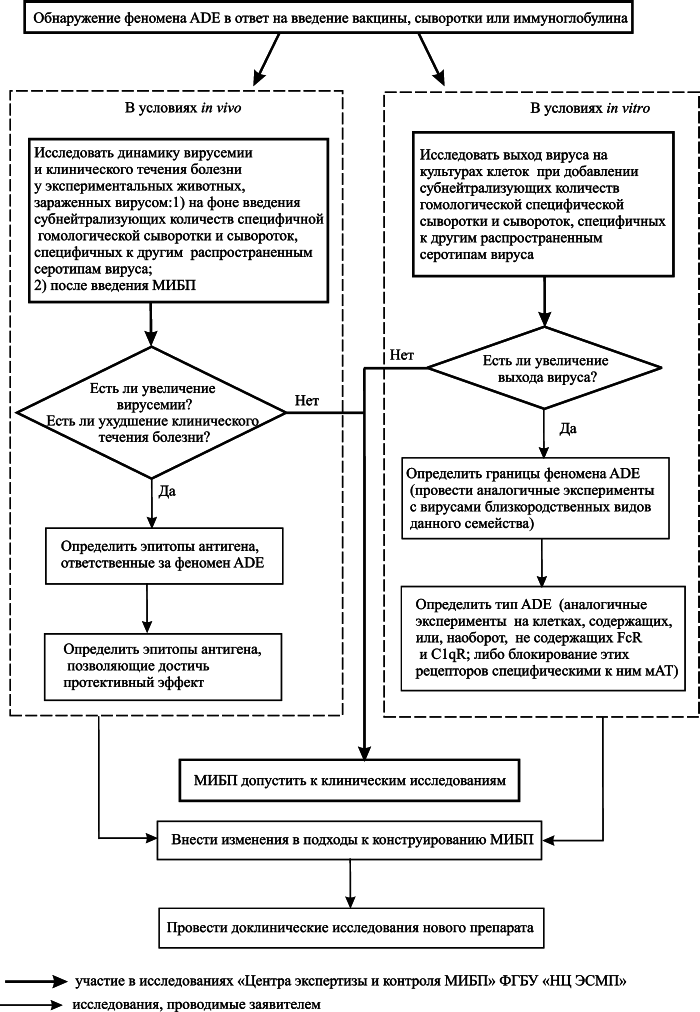
Приведенные выше данные показывают, что не все реакции со стороны иммунной системы, развивающиеся в ответ на введение вакцины или инфекционный процесс, имеют защитный характер. ADE – распространенное явление при инфекционных болезнях и иммунных ответах на введение вакцин и лечение иммуноглобулинами. Осложнения на вакцинацию, связанные с ADE, могут проявляться через десятилетия после ее проведения. Постулат вакцинологии «чем выше титр антител, тем сильнее иммунитет», не может рассматриваться как критерий профилактической эффективности вакцины в тех случаях, когда возможно развитие ADE. Также этот феномен требует по новому взглянуть на подходы к оценке иммунологической безопасности ИЛП. Граждане, которые привлекаются к клиническим исследованиям вакцин, в соответствии с п. 1. ст. 5 Федерального закона № 157-ФЗ от 17.09.1998 г. «Об иммунопрофилактике инфекционных болезней», должны быть информированы о возможности развития у них ADE при заражении возбудителем инфекционной болезни, в отношении которого они были вакцинированы ранее. Поэтому необходима разработка методов исследований, позволяющих еще на этапе доклинического исследования ИЛП устанавливать возможность развития ADE как следствие вакцинации или введения специфических иммуноглобулинов. Ранее нами был предложен алгоритм доклинических исследований, имеющих целью обнаружение ADE при изучении иммунологической безопасности ИЛП (рисунок 7).
Наиболее вероятно развитие ADE у лиц, ранее вакцинированных в отношении вирусов, возбудителей инфекционных болезней, представителей семейств Orthomyxoviridae, Paramyxoviridae, Rhabdoviridae, Coronaviridae, Retroviridae, Parvoviridae, Filoviridae, Flaviviridae, Togaviridae, Picornaviridae, а также возбудителей туберкулеза.
Общими требованиями при доклиническом изучении риска развития ADE у ранее вакцинированных людей, должно быть проведение экспериментов, в которых необходимо установить: 1) границы феномена, т.е. очертить «круг» близкородственных видов вирусов (их серотипов или изолятов), при инфицировании которыми вакцинированных людей возможно усиление инфекционного процесса; 2) тип ADE; 3) эпитопы антигенов, ответственные за феномен ADE, и эпитопы, ответственные за протективный эффект.
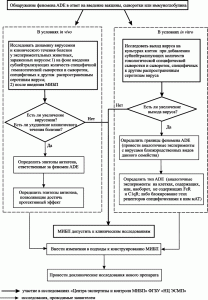 Рис. 7. Блок-схема исследования феномена ADE в доклиническом изучении иммунологической безопасности ИЛП [10]
Рис. 7. Блок-схема исследования феномена ADE в доклиническом изучении иммунологической безопасности ИЛП [10]
Литература
1. Воробьев А.А., Быков А.С., Пашков Е.П. с соавт. Микробиология. М. 1992.
2. Галактионов В.Г. Эволюционная иммунология. М. 2005.
3. Заболотный Д.К. Пустулезная форма чумы // Русский архив патологии. 1899. Т. VIII. С. 239–242.
4. Константинова Н.А. Иммунные комплексы и повреждение тканей. М. 1996.
5. Купер Э. Сравнительная иммунология. М. 1980.
6. Медуницын Н.В. Вакцинология. М., 2010.
7. Павлович С.А. Основы иммунологии. М. 1997.
8. Райт А.Е. Основы вакцинотерапии (теория опсонинов). Спб. 1908.
9. Супотницкий М.В. Эволюционная патология. М. 2009.
10. Супотницкий М.В. Феномен антителозависимого усиления инфекции при доклиническом изучении иммунобиологических лекарственных препаратов // Руководство по проведению доклинических исследований лекарственных средств (иммунобиологические лекарственные препараты). Часть вторая / Под ред. А. Н. Миронова. М.: Гриф и К, 2012. C. 177–185.
11. Baldinotti F. D., Matteucci P., Mazzetti P. et al. Serum neutralization of feline immunodeficiency virus is markedly dependent on passage history of the virus and host system // J. Virol. 1994. V. 74. P. 10834–10837.
12. Balsitis S.J., Williams K.L., Lachica R. et al. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification // PLoS Pathog. 2010. 6:e1000790.
13. Bannert N., Kurth R. Retroelements and the human genome: New perspectives on an old relation // Proc. Natl. Acad. Sci. USA. 2004. V. 101, Suppl. 2. P. 14572–14579.
14. Barrett A. D. T., Gould A. Antibody-mediated early death in vivo after infection with Yellow fever virus // J. Gen. Virol. 1986. V. 67. P. 2539–2542.
15. Blancou J., Andrai B., Andrai L. A model in mice for the study of the early death phenomenon after vaccination and challenge with rabies virus // J. Gen. Virol. 1980. V. 50. P. 433–435.
16. Burke D.S. Human HIV vaccine trials: does antibody-dependent enhancement pose a genuine risk? // Perspect. Biol. Med. 1992. V. 35. P. 511–530.
17. Burton D.R., Stanfield R.L., Wilson I.A. Antibody vs. HIV in a clash of evolutionary titans // Proc. Natl. Acad. Sci. USA. 2005. V. 102, № 42 P. 14943–1494.
18. Chanock R.M., Parrott R.H., Kapakian A.Z. Possible role of immunological factors in pathogenesis of RS virus in lower respiratory tract disease // Perspec. Virol. 1968. V. 6. P. 125–135.
19. Chaturvedi U.C., Raghupathy R., Pasca A.S. et al. Shift from a Th1-type response to Th1-type in dengue haemorrhagic fever // Curr. Sci. 1999. V. 76. P. 63–69.
20. Coffin J. M. Evolution of retroviruses: fossils in our DNA // Proceed. Amer. Philosoph. Soc. 2004. V. 148, № 3. P. 264–280.
21. Costas J., Naverira H. Evolutionary history of the human endogenous retrovirus family ERV9 // Mol. Biol. Evol. 2000. V. 17, № 2. P. 320–330.
22. Dejnirattisai W., Jumnainsong A., Onsirisakul N. Cross-reacting antibodies enhance dengue virus infection in humans // Science. 2010. V. 328. P. 745–748.
23. Delgado M.F., Coviello S., Monsalvo A. C. et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease // Nat. Med. 2009. V. 15. P. 34–41.
24. Dworak L.J., Wolfinbarger J.B., Bloom M.E. Aleutian mink disease parvovirus infection of K562 cells is antibody-dependent and is mediated via an Fc(gamma)RII receptor // Arch. Virol. 1997. V. 142, № 2. P. 363–373.
25. Flipse J., Wilschut J., Smit J.M. Molecular mechanisms involved in antibody-dependent enhancement of Dengue virus infection in humans // Traffic. 2013. V. 14. P. 25–35.
26. Frost S., Wrin T., Smith D.M. et al . Neutralizing antibody responses drive the evolution of human immunodeficiency virus type 1 envelope during recent HIV infection // Proc. Natl. Acad. Sci. USA. 2005. V. 102, № 51. P. 18514–18519.
27. Fulginiti F.A., Eller J.J., Downie A.W., Kempe C.H. Altered reactivity to measles virus. Atypical measles in children previously immunized with inactivated measles virus vaccines //JAMA. 1967. V. 202. P. 1075–1080.
28. Fust G. Enhancing antibodies in HIV infection // Parasitology. 1997. V. 115, Suppl: S 127–140.
29. Goulld E.A., Buckley A. Antibody-dependent enhancement of Yellow Fever and Japanese encephalitis virus neurovirulence // J. Gen. Virol. 1989. V. 70. P. 1605–1608.
30. Halstead S.B., Mahalingam P.S., Marovich M.A. et al. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes // Lancet Infect. Dis. 2010. V. 10, № 10. P. 712–722.
31. Halstead S.B., Chow J., Marchette N.J. Immunologic enhancement of Dengue virus replication // Nat. New Biol. 1973. V. 243. P. 24–26
32. Halstead S.B., Nimmannitya S., Yamarat C., Russell P.K. Hemorrhagic fever in Thailand; recent knowledge regarding etiology // Jpn. J. Med. Sci. Biol. 1967. V. 20 (Suppl.). P. 96–103.
33. Han J.-F., Cao R., Deng Y. et al. Antibody dependent enhancement infection of Enterovirus 71 in vitro and in vivo // Virol. J. 2011. V. 8 .
34. Hawkes R.A. Enhancement of the infectivity of arboviruses by specific antisera produced in domestic fowls // Aust. J. Exp. Biol. Med. Sci. 1964. V. 43. P. 465–482.
35. Hawkes R.A., Lafferty K.J. The enhancement of virus infectivity by antibody // Virology. 1967. V. 33. P. 250–261.
36. He X., Sun X., Wang J. et al. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A // Inf. Immunol. 2009. V. 77, № 6. P. 2294–2303.
37. Henchal E.A., McCown J.M., Burke D.S. et al. Epitopic analysis of antigenic determinants on the surface of Dengue-2 virions using monoclonal antibodies // Am. J. Trop. Med. Hyg. 1985. V. 34. P. 164–169.
38. Homsy J., Meyer M., Tateno M. et al. The Fc and not CD4 receptor mediates antibody enhancement of HIV infection in human cells // Science. 1989. V. 16, № 244. P. 1357–1360.
39. Hosie M.J., Osborne R., Reid G. et al. Enhancement after feline immunodeficiency virus vaccination // Vet. Immunol. Immunopathol. 1992. V. 35. P. 191–197.
40. Huisman W., Karlas K.H., Siebelink K.H. et al. Feline immunodeficiency virus subunit vaccines that induce virus neutralizing antibodies but no protection against challenge infection // Vaccine. 1998. V. 16. P. 181–187.
41. Iankov I. D., Pandey M., Harvey M. et al. Immunoglobulin G antibody-mediated enhancement of measles virus infection can bypass the protective antiviral immune response // J. Virol. 2006. V. 80, № 17. P. 8530–8540.
42. Issel C.J., Horohov D.W., Lea D.F. et al. Efficacy of inactivated whole-virus and subunit vaccines in preventing infection and disease caused by equine infectious anemia virus // J. Virol. 1992. V. 66. P. 3398–3408.
43. Kim H.W., Canchola J.G., Brandt C.D. et al. Respiratory syncytial virus disease in infants despite prior administration of antigenic inactivated vaccine // Am. J. Epidemiol. 1969. V. 89. P. 422–434.
44. King A.A., Sands J.J., Porterfield J.S. Antibody-mediated enhancement of rabies virus infection in a mouse macrophage cell line (P388D1) // J. Gen. Virol. 1984. V. 65. P. 1091–1093.
45. Kreil T.R., Eibl M.M. Pre- and postexposure protection by passive immunoglobulin but no enhancement of infection with a flavivirus in a mouse model // J. Virol. 1997. V. 71, № 4. P. 2921–2927.
46. Kurosu T. Quasispecies of dengue virus // Trop. Med. and Health. 2011. V. 39, № 4. P. 29–36.
47. Lidbury B.A., Mahalingam S. Specific ablation of antiviral gene expression in macrophages by antibody-dependent enhancement of Ross River virus infection // J. Virol. 2000. V. 74. P. 8376–8381.
48. Linn M.L., Aaskov G., Suhrbier A. Antibody-dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis // J. Gen. Virol. 1996. V. 77. P. 407–411.
49. Maglione P.J., Xu J., Casadevall A., Chan J. Fc gamma receptors regulate immune activation and susceptibility during Mycobacterium tuberculosis infection // J. Immunol. 2008. V. 180. P. 3329–3338.
50. Mathew A., West K., Kalayanarooj S. et al. B-cell responses during primary and secondary dengue virus infections in humans // J. Infect. Dis. 2011. V. 204. P. 1514–1522.
51. Mdurvwa E.G., Ogunbiyi P.O., Gakou H.S., Reddy P.G. Pathogenic mechanisms of caprine arthritisencephalitis virus // Vet. Res. Commun. 1994. V. 18. P. 483–490.
52. Mealey R.H., Leib S.R., Littke M.H. et al. Viral load and clinical disease enhancement associated with a lentivirus cytotoxic T lymphocyte vaccine regimen // Vaccine. 2009. V. 27. P. 2453–2468.
53. Mehlhop E., Ansarah-Sobrinho C., Johnson S. et al. C1q Inhibits antibody-dependent enhancement of Flavivirus infection in vitro and in vivo in an IgG subclass specific manner // Cell Host Microbe. 2007 V. 2, № 6. Р. 417–426.
54. Meyer K., Ait-Goughoulte M., Keck Zhen-Yong et al. Antibody-dependent enhancement of hepatitis C virus infection // J. Virol. 2008. V. 82, № 5. P. 2140–2149.
55. Moghaddam A., Olszewska W., Wang B. et al. A potential molecular mechanism for hypersensitivity caused by formalin-inactivated vaccines // Nat. Med. 2006. V. 12. P. 905–907.
56. Nakayama E., Tomabechi D, Matsuno K. et al. Antibody-dependent enhancement of Marburg virus infection // J. Infect. Dis. 2011. V. 204. P. 978–985.
57. Porter A.D., Larsen A.E., Porter H.G. The pathogenesis of Aleutian disease of mink. II. Enhancement of tissue lesions following the administration of a killed virus vaccine or passive antibody // J. Immunol. 1972. V. 109. P. 1–7.
58. Prabhakar B.S., Nathanson N. Acute rabies deaths mediated by antibody // Nature. 1981. V. 290. P. 590–5991.
59. Radkowski M. T., Laskus T., Goch A. et al. Affinity of anti-GP41 antibody in patients infected with human immunodeficiency virus type I // Eur. J. Clin. Invest. 1993. V. 23. P. 455–458.
60. Reed H. Intelligent discussion on HIV vaccine serves as a small consolation for slow progress // Yale J. Biol. 2011. V. 84. P. 153–154.
61. Reimer L.G. Q Fever // Clin. Microbiol. Rev. 1993. V. 6, № 3. P. 193–198.
62. Robinson W.E., Montefiori D.C., Mitchell W.M. Antibody-dependent enhancement of human immunodeficiency virus type 1 infection // Lancet. 1988. V. 9, № 1. P. 790–794.
63. Sekaly R.-P. The failed HIV Merck vaccine study: a step back or a launching point for future vaccine development? // J. Exp. Med. 2008. V. 21. V. 205, № 1. P. 7–12.
64. Shankarappa R., Margolick J.B., Gange S.J. et al. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection // J. Virol. 1999. V. 73, № 12. P. 10489–10502.
65. Shannon J., Heinzen R. Adaptive immunity to the obligate Intracellular pathogen Coxiella burnetii // Immunol. Res. 2009. V. 43, № 1–3. P. 138–148.
66. Suhrbier A., La Linn M. Suppression of antiviral responses by antibody-dependent enhancement of macrophage infection // Trends Immunol. 2003. V. 24. P. 165–168.
67. Takada A., Ebihara H., Feldmann H. et al. Epitopes required for antibody-dependent enhancement of Ebola virus infection // J. Infec. Dis. 2007. V. 196. P. 347–356.
68. Takada A., Feldmann H., Ksiazek T.G. et al. Antibody-dependent enhancement of Ebola virus infection //J. Virol. 2003. V. 77, № 13. P. 7539–7544.
69. Takada A., Watanabe S., Okazak K. et al. Infectivity enhancing antibodies to Ebola virus glycoprotein // J. Virol. 2001. V. 75, № 5. P. 2324–2330.
70. Takano T., Kawakami S., Yamada S. et al. Antibody-dependent enhancement occurs upon re-infection with the identical serotype virus in feline infectious peritonitis virus infection // J. Vet. Med. Sci. 2008. V. 70, № 12. P. 1315–1321.
71. Takeda A., Tuazon C.U., Ennis F.A. Antibody-enhanced infection by HIV-1 via Fc receptor-mediated entry // Science. 1988. V. 242. № 4878. P. 580–5833.
72. Thomas H.I., Wilson S., O'Tolle C.M. et al. Differential maturation of avidity of IgG antibodies to gp41, p24 and p17 following infection with HIV-1 // Clin. Exp. Immunol. 1996. V. 103. P. 185–191.
73. Tirado S.M., Yoon K.J. Antibody-dependent enhancement of virus infection and disease // Viral Immunol. 2003. V. 16. P. 69–86.
74. Tsai Y.T., Chang S.Y., Lee C.N., Kao C.L. Human TLR3 recognizes dengue virus and modulates viral replication in vitro // Cell. Microbiol. 2009. V. 11. P. 604–615.
75. Ubol S., Chareonsirisuthigul T., Kasisith J., Klungthong C. Clinical isolates of Dengue virus with distinctive susceptibility to nitric oxide radical induce differential gene responses in THP-1 cells // Virology. 2008. V. 376. P. 290–296.
76. Ubol S., Halstead S.B. How innate immune mechanisms contribute to antibody-enhanced viral infections // Clin. Vac. Immunol. 2010. V. 17, № 12. P. 1829–1835.
77. Vaccination and Enrollment Are Discontinued in Phase II Trials of Merck's Investigational HIV Vaccine Candidate // News Release .
78. Vennema H., De Groot R., Harbour D. et al. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization // J. Virol. 1990. V. 64. P. 1407–1409.
79. Vennema H., Poland A., Foley J., Pedersen N.C. Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses // Virology. 1998. V. 243, № 1. P. 150–157.
80. Wallace M.J., Smith D.W., Broom A.K. et al. Antibody-dependent enhancement of Murray Valley encephalitis virus virulence in mice // J. Gen. Virol. 2003. V. 84, № 7. P. 1723–1728.
81. Wang S.Z., Rushlow K.E., Issel C.J. et al. Enhancement of EIAV replication and disease by immunization with a baculovirus-expressed recombinant envelope surface glycoprotein // Virol. 1994. V. 199. P. 247–251.
82. Webster R.G., Askonas B.A. Cross-protection and cross reactive cytotoxic T cells induced by influenza virus vaccines in mice // Eur. J. Immunol. 1980. V. 10. P. 396–401.
83. Weiss R.C., Scott F.W. Antibody-mediated enhancement of disease in feline infectious peritonitis: comparisons with dengue hemorrhagic fever // Comp. Immune. Microbiol. Infect. Dis. 1981. V. 4. P. 175–188.
84. Yoon K.J., Wu L.L., Zimmerman J.J., Hill H.T., Platt K.B. Antibody-dependent enhancement (ADE) of porcine reproductive and respiratory syndrome virus (PRRSV) infection in pigs // Viral. Immunol. 1996. V. 9. P. 51–63.
85. Yoong P. Enhancement of bacterial virulence by antibody neutralization of immune-activating toxins // Virulence. 2010. V. 1, № 5. P. 409–413.
86. Zellweger R.M., Prestwood T.R., Shresta S. Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease // Cell. Host. Microbe. 2010. Vol. 7. P. 128–139.
87. Zhang H., Hoffmann F., He J. et al. Evolution of subtype C HIV-1 Env in a slowly progressing Zambian infant // Retrovirology. 2005 (http://www.retrovirology.com/content/2/1/6).
Библиографическое описание: Миронов А.Н., Супотницкий М.В. Лебединская Е. В. Феномен антитело-зависимого усиления инфекции у вакцинированных и переболевших // Биопрепараты. 2013. № 3. С.12–25.
Bibliographical description: Mironov A.N., Supotnitskiy М.V., Lebedinskaya E.V. The phenomenon of antibody-dependent enhancement of infection in vaccinated and convalescents // Biopreparats (Biopharmaceuticals). 2013. No. 3. P. 12–25.
ССЫЛКИ ПО ТЕМЕ:
Супотницкий М.В. Неисследованные тупики вакцинации // Крымский журнал экспериментальной и клинической медицины. — 2011. — Т. 1, № 3–4. — С. 118–127.
Супотнiцький М.В. Чому ми не здолаемо ВIЛ/СНIД (Почему мы не победим ВИЧ/СПИД-пандемию) // Iнфекцiйнi хворобию. — 2012. — № 1 (67). — С. 88–96; № 2 (68). — С. 104–114.
Супотницкий М.В. Антитела в инфекционных и эпидемических процессах // Новости медицины и фармации. — 2013. — № 8 (456).
[1] Т-хелперы 1 (Th1) – преимущественно способствуют развитию клеточного иммунного ответа, активируя Т-киллеры; Т-хелперы 2 (Th2) – активируют В-лимфоциты, способствуя развитию гуморального иммунного ответа.
[2] Fc-фрагмент Ig (кристаллизующийся фрагмент иммуноглобулина, fragment crystallizable region, Fc region) – концевая часть молекулы иммуноглобулина, которая взаимодействует с Fc-рецептором на поверхности клетки и с некоторыми белками системы комплемента. Другая часть антитела называется Fab (от англ. Fragment antigen binding), и состоит из двух вариабельных участков, определяющих специфичность мишени, которую связывает антитело. Fc-фрагменты антител одного класса консервативны.
[3] Fc-рецепторы (Fc receptors) – представляют собой семейство молекул, каждый член которого распознает иммуноглобулин одного или нескольких родственных изотипов. Рецепторы этого типа входят в состав суперсемейства иммуноглобулинов. Fc-рецепторы для иммуноглобулинов присутствуют на поверхности мононуклеарных лейкоцитов, нейтрофилов, нормальных клеток-киллеров, эозинофилов, базофилов и тучных клеток. Взаимодействуя с Fc-областью иммуноглобулинов разных изотипов, эти рецепторы стимулируют, например, фагоцитоз, противоопухолевую цитотоксическую активность и дегрануляцию тучных клеток.
[4] Этот процесс отличается от образования иммунных комплексов, приводящих к развитию так называемых иммунокомплексных болезней (ревматоидный артрит, сывороточная болезнь, системная красная волчанка и др.), тем, что комплексы «вирус–специфическое антитело» не вызывают прямого повреждения тканей и органов. Их патологическое действие проявляется через усиление инфекционного процесса. Более подробно с патогенезом иммунокомплексных болезней можно ознакомиться по монографии Н.А. Константиновой [4].
[5] С1 – компонент комплемента. Его субъединица C1q имеет рецептор для связывания с Fc-фрагментом молекулы антитела.
[6] Моноциты образуются в костном мозге из гемопоэтических стволовых клеток-предшественников. Они циркулируют в кровяном русле 1–3 сут, затем созревают в резидентные макрофаги и дендритные клетки. Моноциты – наиболее активные фагоцитирующие клетки периферической крови.
[7] (+)ssРНК означает, что вирус содержит одноцепочечную (single-stranded, ss) плюс-цепь РНК, которая используется им в качестве мРНК и генома.
[8] Toll-подобный рецептор – рецептор системы иммунитета, подобный рецепторному белку Toll плодовой мушки (Drosophila).
[9] Геликазы – ферменты, расплетающие двухцепочечные ДНК или РНК у вирусов, бактерий и эукариот.
[10] Более подробно феномен первичного антигенного греха при инфекционных процессах будет рассмотрен в следующем сообщении.
[11] Однако в слюнных железах переносчиков DENV – комаров, гетерогенизация вируса возобновляется [46]. Мы объясняем это тем, что у членистоногих хорошо развита «питательная среда» для вируса – фагоцитарная система, однако для гуморальных факторов их иммунной системы характерно отсутствие высокой специфичности, присущей антителам позвоночных (см. в работе [5]).
[12] Подробное описание iADE при лихорадке Денге можно найти в работах [25, 76].
[13] В России такая вакцина не используется.

Михаил Васильевич Супотницкий
Российский микробиолог, полковник медицинской службы запаса, изобретатель, автор книг и статей по истории эпидемий чумы и других особо опасных инфекций, истории разработки и применения химического и биологического оружия. Заместитель главного редактора научно-практического журнала «Вестник войск РХБ защиты» Министерства обороны РФ.